Причины мышечной дистрофии Эрба-Рота
Причиной развития данной патологии является генетический дефект в гене 13q12, 17q12-q21.33, 4q12 и 5q33. Это обозначает, что в мышечной клетке образуется недостаточное количество ферментов. Именно они нужны клетки для последующего строительства белка. Выходит, что по причине недостаточности белка, повышается проницаемость мембран клеток. То есть, синтез саркогликанов нарушен, поэтому и нарушен белковый комплекс дистрофин-гликопротеин.
Белковый комплекс дистрофин-гликопротеин обеспечивает связь клеточного скелета сократительных элементов мышечных волокон миофибрилл с внеклеточными тканевыми структурами. В результате дефицита саркогликанов баланс аминокислот и ферментов в мышечных волокнах нарушается. Каркас мышечной клетки создает белок дистрофин. Каркас мышечной клетки похож на сетку Рабицу. Еще точнее на соты с медом. Если в одной соте меда не будет, то соседние соты увеличиваются в размере и немного сдвигаются одной стенкой в место пустой соты. Мед из увеличенных сот начинает вытекать.
Рис. 1. Генетический дефект при дистрофии Эрба-Рота
Точно так же начинает вытекать их мышечной клетки фермент креатинфосфокиназа. Он вытекает из клетки и попадает сначала в лимфу, затем из лимфы в кровь. Наличие в крови повышенного содержания креатинфосфокиназы показывает, что мышечная клетка в беде, т.е в ней нарушены биохимические реакции.
Для чего нужна креатинфосфокиназа мышечной клетки в достаточном количестве? Креатинфосфокиназа нужна мышечной клетки для того, чтобы митохондрия могла создавать энергию. Энергию в мышечной клетке создает митохондрия.
Мышечная слабость при мышечной дистрофии Эрба-Рота создает недостаточное количество креатинфосфокиназы внутри мышечной клетки.
Типы поясно-конечностной мышечной дистрофии
Ниже представлен список типов ПКМД.
Для типа 1 характерно наследование по доминантному признаку, т.е. требуется только одна мутация для проявления заболевания.
Для типа 2 характерно наследование по рецессивному признаку, т.е. требуется две мутации в гене – по одной от каждого родителя.
Некоторым типам ПКМД вместо чисел присвоены названия.
Типы ПКМД по отдельным названиям:
- миопатия Бетлема (мутация в гене collagen 6, доминантная)
- кальпаинопатия (мутация в гене calpain, рецессивная, другое название — LGMD2A)
- дисферлинопатия (мутация в гене dysferlin, рецессивная, другое название – LGMD2B)
- миофибриллярная миопатия (мутации в генах desmin, alpha-B crystallin, myotilin, ZASP, filamin C, BAG3 или SEPN1; все доминантные кроме desmin-типа, который может быть как доминантным, так и рецессивным)
- саркогликанопатии (мутация в гене sarcoglycan; рецессивная; другие названия — LGMD2C, LGMD2D, LGMD2E, LGMD2F)
- ZASP-связанные миопатии (мутация в гене ZASP; доминантная; является формой миофибриллярной миопатии)
Доминантные типы ПКМД по номерам:
- LGMD1A / ПКМД1А (мутация в гене myotilin)
- LGMD1B / ПКМД1B (мутация в гене lamin A/C)
- LGMD1C / ПКМД1С (мутация в гене caveolin)
- LGMD1D / ПКМД1D (мутация в гене DNAJB6)
- LGMD1E / ПКМД1E, также называемая десминовая миопатия – тип миофибриллярной миопатии (мутация в гене desmin)
- LGMD1F / ПКМД1F (мутация на 7-й хромосоме)
- LGMD1G / ПКМД1G (мутация на 4-й хромосоме)
- LGMD1H / ПКМД1H (мутация на 3-й хромосоме)
Рецессивные типы ПКМД по номерам:
- LGMD2A / ПКМД2A (мутация в гене calpain)
- LGMD2B / ПКМД2B (мутация в гене dysferlin)
- LGMD2C / ПКМД2C, также называется SCARMD1 (мутация в гене gamma sarcoglycan)
- LGMD2D / ПКМД2D, также называется SCARMD2 (мутация в гене alpha sarcoglycan)
- LGMD2E / ПКМД2E (мутация в гене beta sarcoglycan)
- LGMD2F / ПКМД2F (мутация в гене delta sarcoglycan)
- LGMD2G / ПКМД2G (мутация в гене telethonin)
- LGMD2H / ПКМД2H (мутация в гене TRIM32)
- LGMD2I / ПКМД2I (мутация в гене FKRP)
- LGMD2J / ПКМД2J (мутация в гене titin)
- LGMD2K / ПКМД2K (мутация в гене POMT1)
- LGMD2L / ПКМД2L (мутация в гене ANO5)
- LGMD2M / ПКМД2M (мутация в гене fukutin)
- LGMD2N / ПКМД2N (мутация в гене POMT2)
- LGMD2O / ПКМД2O (мутация в гене POMGnT1)
- LGMD2Q / ПКМД2Q (мутация в гене plectin)
Источник Muscular Dystrophy Association
Что делает мышечная клетка, чтобы остановить выход креатинфосфокиназы наружу?
Чтобы сохранить нужные вещества, клетка вынуждена закрывать эти дыры. А их можно закрыть только теми веществами, которые больше этих дыр. Мышечная клетка начинает задерживать внутри себя жировые компоненты, которые больше этих дыр.
Чтобы удержать жировые компоненты около этих дыр, нужно приложить силу. Чтобы приложить силу, нужна энергия. Энергию создают митохондрии. Поэтому, чтобы спасти жизнь клетки и свою жизнь, митохондрии перемещаются от двигательных белков актина и миозина к стенкам, полу и потолку клетки. Актин и миозин остаются без части энергии. Клетка, перепуганная тем, что могут образовываться другие дыры в каркасе, озабочена тем, что создает внутри себя дополнительные жировые включения (на всякий случай). Этих включений становиться так много, что жир начинает зажимать двигательные белки клетки до полного их обездвиживания.
Рис. 2. Состояние мышечной клетки при мышечной дистрофии Эрба-Рота
Пояснение к фото:
A — изменение размеров мышечных волокон и некротических миофибрилл (стрелки).
B — кластер базофильных регенерирующих миофибрилл (стрелки).
C — иммуногистохимическое окрашивание дистрофина демонстрирует заметную потерю нормальной окраски плазматической мембраны.
Как действует мой метод для восстановления нормального движения мышц?
Применяя мой метод, Никонова Николая, при дистрофии Эрба-Рота: растяжения мышцы, фиксация ее в определенном положении и надавливая на нее определенным техническим приемом, происходит освобождение двигательных белков от пресса жировых включений.
В электронный микроскоп я вижу увеличение присутствия жировых клеток и концентрацию митохондрий у стенок клеток, что подтверждает мои логические рассуждения.
Рис. 3. Биопсия мышцы при диагнозе мышечная дистрофия Эрба-Рота
Биопсия мышцы показывает выраженный фиброз эндомизиального фибиса (A) и инфильтрацию лимфоцитов (B). (C) показаны базофильные регенерирующие волокна. (D) Некротический миофиб инфильтрирован отмеченными лимфоцитами и гистиоцитами
До тех пор, пока жировые включения, своей тяжестью, не остановили работу двигательных белков, движение мышц есть. Пусть слабые, пусть быстро утомляемые, но движения мышц есть.
Как только жировые включения зажимают двигательные белки, то образуется обездвиживание. Если обездвиживание произошло в диафрагме, то останавливается дыхание. Если обездвиживание произошло в сердце, то останавливается сердце.
Восстановить работу гена я не могу, но …, воздействуя на мышцы моим методом, получилось уменьшить количество жировых компонентов внутри мышечной клетки у девочки с прогрессирующей мышечной дистрофией Эрба-Рота и часть митохондрий вернулось на свои места.
Мышцы стали сокращаться и дали возможность Насте начать передвигаться.
Сейчас Настя находиться у себя дома, ухудшения ее состояния нет. Даже есть некоторое улучшения – Настя стала плавать в бассейне.
Доктор Никонов
Неудобство в восстановлении мышечной дистрофии Эрба-Рота в том, что нужно постоянно периодически воздействовать на мышцы моим методом, чтобы не было чрезмерно больших жировых отложений в мышечных клетках.
При сахарном диабете люди всю жизнь испытывают неудобства, принимая инсулин, но несмотря на это живут полноценной жизнью (заводят семью и детей).
Горжусь моими знаниями, опытом и навыками!
Результат моих знаний по восстановлению нормальной работы мышц при мышечной дистрофии Дюшенна:
Мышечная сила появилась у Эмине, Сергея, Якоба. Они и другие мои пациенты ходят, как обычные здоровые люди!
Симптомы при дистрофии Эрба-Рота
Привожу ключевые симптомы при данном диагнозе, который начинает развиваться у детей и подростков:
- Происходит задержка в начале самостоятельно ходьбы ребенка.
- Неудобная для больного походка, которая выглядит как ковыляние с ноги на ногу. Это еще называют как “утиный” тип ходьбы. Это происходит из-за симметричного ослабления мышц области беда.
- Ребенок часто спотыкается при передвижении и падает при беге, другими словам – дисбаланс и нестабильность.
- Сложности, возникающие при попытке встать с постели, стула. Также проявляются трудности при ходьбе на склонах, восхождениях и даже при спуске по лестнице.
- Наблюдается выпуклость лопаточных костей. Это происходит по причине ослабления передних зубчатых мышц грудной клетки больного и ромбовидных мышц спины.
- Уменьшается окружность талии. Это связано с тем, что при мышечной дистрофии Эрба-Рота происходит уменьшение тонуса поперечных мышц грудной клетки, живота и ileal-rib.
- Патологическая усталость у ребенка.
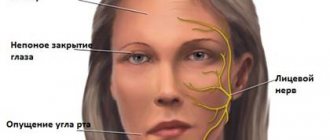
В результате прогрессирования болезни наблюдается постоянная общая слабость и ослабление мышечного корсета спины и мышц плечевого пояса. Эти процессы приводят к таким дефектам осанки как гиперлордоз. Пациентам с дистрофией становиться с каждым разом все труднее, как и удерживать предметы в руках, поднимать сами руки к верху. Что касается мимических лицевых мышц, то они тоже теряют свою подвижность. Это приводит к том, что наблюдается неполное закрытие век и выступ губ.
Процесс постепенного уменьшения мышечного тонуса приводит к неизбежному истончению и дряблости мышечной ткани больного на прогрессирующую дистрофию Эрба-Рота, заменяя ее жировой и фиброзной тканью, т.е. миодистрофией.
Симптомы заболевания на поздних стадиях
Серьезная потеря мышечной массы, сгибательная контура, сокращение сухожилий у больного и практически полная потеря глубоких сухожильных рефлексов нижних конечностей ребенка (колено и подошва).
Диагностика мышечной дистрофии Эрба-Рота
- Основывается диагностика рассматриваемого заболевания на физическом обследовании больных, исследовании семейной истории пациента и последующем анализе собранных данных.
- Проводится генетическое тестирование. Нужно для последующего точно определения мышечной дистрофии.
- Электронейромиография.
- Проводится биопсия мышечной ткани с биохимическим исследованием.
- Сдается общий анализ крови.
- Сдается анализ крови на креатинфосфокиназа.
- Анализ мочи больного.
Что касается электромиографии, то она позволяет исследовать не только степень нервно-мышечной передачи, но и определить уровень непосредственной мышечной возбудимости, что крайне важно для дифференциальной диагностики заболевания с патологиями нейропатических мышц.
Что такое поясно-конечностная мышечная дистрофия?
Примечание переводчика. В разных источниках название варьируется: поясно-конечностная МД и конечностно-поясная МД.
Поясно-конечностная мышечная дистрофия (ПКМД) – это не одно общее заболевание. Это целая группа заболеваний, поражающих мышцы, в основном расположенные в районе бёдер и плеч.
Плечевой пояс – это костная структура, которая окружает плечевую область.
(Информация из Википедии: плечевой пояс (пояс верхних конечностей) — совокупность костей (пары лопаток и ключиц) и мышц, обеспечивающих опору и движение верхних (передних) конечностей.)
Тазовый пояс – это костная структура, окружающая район бёдер.
Совокупно они называются «пояса конечностей». При ПКМД больше всего повреждаются мышцы, соединённые с костями этих поясов.
Термин «проксимальный» также используется для описания повреждённых при ПКМД мышц. Проксимальные мышцы – это мышцы, расположенные близко к центру тела. Дистальные мышцы – это более удалённые от центра тела мышцы (например, мышцы кистей или ступней). Дистальные мышцы при ПКМД поражаются на поздних этапах, но могут и сохранить свою функцию.
По данным на конец 2012 года насчитывается более 20 различных подтипов ПКМД. Это сложная и постоянно развивающаяся область исследований.
Лечение дистрофии Эрба-Рота
Следует сразу же отметить, что поврежденный ген я не могу восстановить, но…
Доктор Никонов
Воздействие по моему методу направлено на снижение интенсивности симптомов, замедление прогрессирования заболевания, повышение силы в мышцах, восстановление правильного движения во всех группах мышц.
Я не занимаюсь лечением мышечной дистрофии Эрба-Рота. Я занимаюсь восстановлением нормальной работы мышц. Поэтому лечебные процедуры, применяемые в стационарах и в других реабилитационных центрах, описывать в моей статье не буду. Скорее всего, вы их испробовали.
Моя статья носит ознакомительный характер. Информацию я взял из своих наблюдений и десятилетних изучений-открытий ученых со всего мира.
Медицинская справка
Заболевание представляет собой полиморфный вариант наследственной миодистрофии. От других разновидностей патологии оно отличается клинической картиной, течением и временем начала. Впервые описание недуга представил немецкий невролог В. Эрб в 1882 году. Одновременно с ним этой проблемой в России занимался В. Рот, которую позднее он обозначил как «мышечная сухотка». Именно по фамилиям двух ученых и было названо заболевание. В современной неврологии используется несколько его наименований — прогрессирующая мышечная дистрофия Эрба-Рота, конечностно-поясная мышечная дистрофия.
Патология начинает свое развитие, как правило, в детстве. Однако возраст появления первых симптомов может колебаться в диапазоне от 10 от 30 лет. Мужчины и женщины в равной степени страдают от проявлений мышечной дистрофии. Неврологи отмечают, что начавшийся в детстве недуг быстро прогрессирует, если сравнивать его течение в подростковом и взрослом возрасте. Кроме того, во втором случае он протекает в легкой форме.