О других заболеваниях на букву «Б»: Базилярная импрессия, Базилярная мигрень, Бери-бери, Беттолепсия, Боковой амиотрофический склероз, Болезнь Альцгеймера, Болезнь Вильсона, Болезнь Галлервордена-Шпатца, Болезнь Гамсторпа, Болезнь Гиппеля-Линдау, Болезнь Канавана, Болезнь Крейтцфельдта-Якоба, Болезнь Лафоры, Болезнь Мачадо-Джозефа, Болезнь мойя-мойя, Болезнь Мортона, Болезнь Паркинсона, Болезнь Пика, Болезнь Рефсума, Болезнь Фара.
Заболевание Галлервордена-Шпатца — прогрессирующее унаследованное расстройство, которое бывает ввиду отложений железа в головном мозге. Выражается болезнью Паркинсона, сбоем в умственном восприятии и в состоянии сознания, проявлением судорожного симптома, сокращением групп мышц, зрительными нарушениями.
Важным признаком наличия данной патологии является рисунок «глаза тигра» в виде округлой или линейной зоны в переднем мозге, который видно при проведении магнитно-резонансной томографии.
Терапия направлена на устранение симптоматики болезни средствами, применяемыми для лечения двигательных расстройств: препаратами для увеличения уровня домофина, медикаментами против эпилепсии. Также лечение направлено на остановку судорог и подавление депрессивного состояния пациента. В наше время заболевание неизлечимо и все действия направлены исключительно на ослабление негативных проявлений.
Заболеванию Галлервордена-Шпатца была дана характеристика в 1922 году немецкими специалистами по исследованию головного мозга, именно по фамилиям этих морфологов и была названа болезнь. Наиболее частыми клиническими проявлениями этой аномалии считаются совокупные симптомы, которые проявляются в конвульсиях, сокращениях мышц и тиках. Также отличительной чертой являются признаки паркинсонизма, умственного отклонения, патология зрительных нервов, поражение сетчатой оболочки глаза.
Данная патология встречается достаточно редко. После развития выраженных явлений патологии после бессимптомного течения отмечают три формы: детская, юношеская и взрослая.
До появления необходимой аппаратуры диагноз ставился исключительно специалистами по направлению «патологическая анатомия». После внедрения в медицинские организации необходимого оборудования появилась возможность ставить диагноз при жизни. О причине накопления железа стало известно лишь в 2001 году. Выяснилось, что причиной всему служит повреждение гена, который и вызывает нарушение работы головного мозга. После результатов данных исследований заболевание было переименовано в справочниках в пантотенаткиназа-ассоциированную нейродегенерацию.
Причины развития болезни
Заболевание Галлервордена-Шпатца появляется в связи с нарушением работы генов. Оно бывает и наследственным, и случайно приобретенным вследствие определенных мутаций. Генетической основой считают отклонение в гене пантотенаткиназы. Генетическим субстратом выступают нарушения в гене ферментов фотосинтеза, обозначенном на карте 20-й хромосомы 20 р 12.3 – р 13.
Выявлено порядка 50 мутаций. В конечном итоге такое генетическое повреждение приводит к уменьшению пантотенаткиназы, что ведет к сосредоточению серосодержащей кислоты. Данная аминокислота устанавливает прочное соединение с железом и пагубно воздействует на белки. В результате реакции происходит окисление, которое программирует гибель клетки. На месте разрушенных клеток происходит размножение глиальных клеток. Такая необъяснимая реакция чаще всего затрагивает парную структуру переднего мозга и черное вещество, расположенное в области среднего мозга. Именно там и находят сконцентрированное железо, которое окрашено в коричневый цвет. Так же в результате исследований были найдены образования, которые находились в мозговом веществе, коре мозга и в периферической нервной системе.
Особенности генетического сбоя
Болезнь Галлервордена-Шпатца это наследственная генетическая патология. Заболевание может быть как семейным, так и спорадическим (случайным), однако всегда передается по наследству. Болезнь относится к аутносомно-рецессивному типу наследования.
Для передачи болезни оба родителя должны быть гетерозиготными носителями заболевания и иметь одну мутировавшую аллель.
Болезнь вызывается мутацией в гене пантотенат-киназы (PANK2) который находится в 20 хромосоме в локусе 20р12.3–р13. Ген пантотенат-киназы отвечает за кодирование протеина пантотенат-киназы 2, который в свою очередь сдерживает процесс накопления аминокислоты цистеина и пантетеина.
В результате происходит образование химических соединений с ионами железа, которые имеют негативное воздействие на белки и дают старт процессам перекисного окисления, что приводит к программируемой клеточной гибели нейронов. Вместо отмерших нейронов нарастает глиальная ткань (вспомогательные клетки нервной ткани).
Патологические процессы происходят в бледном шаре, красных ядрах и черном веществе (субстанции nigra). В них скапливается субстанция зеленовато-коричневого цвета, которая содержит железо. Помимо этого в белом веществе головного мозга, коре головного мозга, периферических нервных стволах и спинном мозге наблюдаются сфероидные нейроаксональные образования (расширения аксонов с разрастанием тубулярных и мембранных структур).
Симптомы болезни
Самая известная разновидность заболевания Галлервордена-Шпатца начинается в детстве в период с 4 до 10 лет. Самыми распространенными признаками болезни считаются:
Проблемы с мышечным тонусом, особенно ног. Также случается, что больной занимает позы, которые выглядят неестественно. В таких случаях ребенку трудно ходить.- Затем процесс затрагивает туловище, лицо и глотку.
- Так же с вышеуказанными проявлениями бывает дистония, которая наблюдается в нескольких частях тела. Бывает и появляется локальная дистония, непроизвольный спазм мышцы глаза, лицевой спазм, спастическая кривошея.
- Повышенный тонус мышц, при котором мышечная ткань становится плотной и возникает состояние малой двигательной активности с ограничением количества движений.
- Эписиндром.
- Психические нарушения, такие как снижение памяти и внимательности. Бывает в дальнейшем развивается умственная отсталость, именуемая олигофренией. Поведение таких пациентов может стать агрессивными и отклоняться от морально-нравственных норм.
- Нарушение произношения и зрения в результате атрофии нервов можно выделить как самостоятельный симптом у некоторых пациентов.
Диагноз, поставленный в детском возрасте, обычно через 10-15 лет ведет к полной обездвиженности.
Юношеская форма патологии Галлервордена-Шпатца появляется в возрасте 10 лет и имеет замедленное течение. Также возникают проблемы с тонусом мышц, появляются непроизвольные сокращения, которые локализуются в конечностях, мышцах рта и челюсти. Присутствуют и типичные поведенческие особенности, связанные с интеллектом и психикой.
Взрослый вид недуга Галлервордена-Шпатца устанавливается уже после совершеннолетия. Выражается также неврологическими симптомами, которые характеризуются тремором и мышечным тонусом. Выявляется недостаточная двигательная активность, болезненное состояние мышц, тремор и потеря координации движений. Потерю координации легко можно выявить с помощью пробы Тавенарда — стоящего больного подталкивают вперед, тем самым выводя из равновесия. Врач обычно находится за спиной.
Главной особенностью течения заболевания считается совокупность неврологических признаков с другими отклонениями группы мышц. Категория нарушений умственных процессов может варьироваться от сохранности всех функций до прогрессирующего слабоумия.
Особенности клинической картины
Признаки данной патологии могут варьироваться в каждом конкретном случае, и проявление симптомов зависит от формы болезни у индивида.
Детская форма Галлервордена Шпатца проявляется в возрасте между 5 и 10 годами. Считается классической формой болезни.
В подавляющем количестве случаев (90%) заболевание начинается с торсионной дистонии, которая поражает мышцы ног. У больного происходит сокращение мышц, затрудняется ходьба, изменяется походка. Затем поражаются мышцы лица, глотки и туловища.
Возможно присутствие блефароспазма, спазма кистей, спастической кривошеи, лицевого гемиспазма. Треть пациентов имеют мышечную ригидность, гипокинезию, тремор (признаки синдрома паркинсонизма).
К типичным признакам детской формы болезни также относят:
- эпилептический синдром;
- агрессивность;
- умственную отсталость (возникает из-за ухудшения памяти и внимания);
- асоциальность;
- зрительные нарушения (атрофия зрительных нервов, дегенерация сетчатки);
- психические расстройства.
Детская форма заболевания прогрессирует быстро, и полная потеря способности к передвижению наступает через 10-15 лет после дебюта болезни.
Подростковая форма развивается и протекает более медленно. В начале болезни проявляются:
- фокальная торсионная дистония (чаще всего затрагивает мышцы конечностей и челюстно-ротовые мышцы);
- нейропсихологические расстройства;
- поведенческие расстройства;
- интеллектуальные расстройства.
Атипичная форма болезни – наиболее редкая (15% случаев заболевания), и протекает отлично от первых двух форм. Для этой формы наиболее характерны такие симптомы:
- синдром паркинсонизма с невозможностью сохранять равновесие в положении стоя;
- непроизвольные движения в разных частях тела, дистония, хорея и атетоз (замедленные и порывистые движения), гемибаллизм (размашистые движения), миоклония (не длительные мышечные спазмы);
- деменция;
- эпилептический синдром;
- депрессивность, агрессия;
- патологические рефлексы, которые возникают вследствие разрушительных процессов в нейронах головного мозга.
Диагностика
Вследствие проявления различных симптомов неврологу установить точный диагноз заболевания Галлервордена-Шпатца не всегда представляется возможным. Доминирующим признаком считается возрастной фактор до 30 лет, комплекс нарушений двигательной активности, увеличение количества симптомов, а также наличие на изображениях МРТ характерного признака.
Вспомогательными признаками данного заболевания могут служить патологические рефлексы, снижение умственных способностей, эписиндром, ухудшение зрения, атрофия сетчатки глаза, а также наличие в роду данного заболевания.
В целях диагностики проводят дополнительные исследования, в частности делают пациенту электроэнцефалограмму. Для постановки диагноза опираются на данные неврологического статуса и обследования, которое заключается в закономерности суммарной электрической активности мозга.
При явном ухудшении зрения требуется помощь офтальмолога для постановки диагноза, который проведет исследование по определению остроты зрения и осмотрит глазное дно специальными инструментами.
Генетик определяет наличие мутаций при составлении генеалогического древа, а также делает диагностику ДНК. Также с помощью дополнительного проведения ПЭТ выявляют сниженный обмен веществ в образовании серого вещества подкорковой части головного мозга.
Существует ряд других болезней со схожими патологиями, но в их клиническую картину не входит ряд особенностей заболевания Галлервордена-Шпатца. Схожие болезни по симптоматике: болезнь Хантингтона, Вильсона, Мачадо-Джозефа и другие болезни, связанные с генетически поражениями головного мозга.
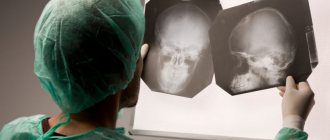
Врачи при постановке диагноза основываются на данные, полученные в результате МРТ. На всех снимках пациентов наблюдается пигментированная область с железом. Изображение с однозначным видом данной болезни получило названия «Глаз тигра».
Более темные ткани этой зоны выглядят как глаз, а более светлые, яркие как зрачок. Момент появления «Глаза тигра» еще не определен учеными. По мнению одних он появляется еще до определенной симптоматики, по мнению остальных через некоторое время после яркого проявления болезни.
Диагностические критерии и методики
Постановка диагноза «болезнь Галлервордена-Шпатца» может быть проблематичной для неврологов из-за полиморфизма симптомов.
Основные критерии для диагностики:
- начало болезни до 30 лет;
- типичная картина в результатах МРТ;
- постоянное развитие симптомов;
- экстрапирамидальные синдромы;
- пирамидные знаки;
- эпилептические приступы;
- когнитивные нарушения;
- пигментная дегенерация сетчатки;
- наличие болезни в семейном анамнезе.
«Глаза тигра» на МРТ с контрастом — типичная картина при болезни Галлервордена Шпатца
Болезнь необходимо дифференцировать с другими заболеваниями, которые имеют сходную клиническую картину: хореей Гентингтона, болезнью Вильсона-Коновалова, хореоакантоцитозом, болезнью Мачадо-Джозефа.
Для диагностирования пациент направляют на такие обследования:
- Изучение неврологического статуса у невролога, невропатолога.
- Электроэнцефалография.
- Консультация офтальмолога. Возможно проведение прямой офтальмоскопии, проверки остроты зрения.
- Генетические исследования, для определения типа наследования.
- Исследования ДНК (обнаружение мутаций в гене PANK2).
- Позитронная эмиссионная томография головного мозга. Проводится для обнаружения снижения кровообращения в лобно-теменных долях, стриатуме, бледном шаре.
- Магнитно-резонансная томография головного мозга. Требуется для обнаружения «глаза тигра» — гиперинтенсивного овального участка в гипоинтенсивной зоне, который возникает из-за скопления железа. Не существует единой теории о развитии «глаза тигра», есть предположения о его возникновении до начала клинических проявлений болезни, и о его появлении через годы после начала течения заболевания. Также МРТ позволяет обнаружить сфероидные нейроаксональные образования.
Протекание заболевания
Обычное начало заболевания в детском возрасте, иногда с выраженной симптоматикой уже на первом году жизни. Редко возможна манифестация во взрослом возрасте. Сначала возникают экстрапирамидные моторные нарушения, в особенности нарушения ходьбы с тенденцией к падениям или дистонией ног, реже — психические нарушения. В дальнейшем — нарушения движений (дистонии, хореоатетоз, тремор) с ригидностью затылочной мускулатуры, гиперрефлексией и нарушениями психики (как правило, расстройство интеллекта в виде прогрессирующей деменции). Также обнаруживаются дизартрия и дисфагия. Можно обнаружить пигментацию сетчатки или атрофию зрительного нерва. У взрослых преобладает синдром паркинсонизма-плюс — с деменцией, гиперрефлексией и проминентной дистонией. Течение заболевание прогрессивно, неврологическое состояние пациентов ухудшается постепенно.
Смерть наступает обычно в возрасте до 30 лет, в состоянии полного психического распада личности.