Causes of Erb-Roth muscular dystrophy
The cause of the development of this pathology is a genetic defect in the gene 13q12, 17q12-q21.33, 4q12 and 5q33. This means that an insufficient amount of enzymes is formed in the muscle cell. These are exactly what cells need for subsequent protein construction. It turns out that due to protein deficiency, the permeability of cell membranes increases. That is, the synthesis of sarcoglycans is impaired, and therefore the dystrophin-glycoprotein protein complex is impaired.
The dystrophin-glycoprotein protein complex ensures the connection of the cellular skeleton of the contractile elements of muscle fibers of myofibrils with extracellular tissue structures. As a result of sarcoglycan deficiency, the balance of amino acids and enzymes in muscle fibers is disrupted. The muscle cell framework creates the protein dystrophin. The framework of a muscle cell is similar to a chain-link mesh. Even more precisely, on honeycombs with honey. If there is no honey in one comb, then the neighboring combs increase in size and move slightly with one wall in place of the empty comb. Honey begins to flow out of the enlarged honeycombs.
Rice. 1. Genetic defect in Erb-Roth dystrophy
In the same way, the enzyme creatine phosphokinase begins to leak out of their muscle cells. It flows out of the cell and enters first the lymph, then from the lymph into the blood. The presence of increased levels of creatine phosphokinase in the blood indicates that the muscle cell is in trouble, that is, its biochemical reactions are disturbed.
Why is creatine phosphokinase needed in muscle cells in sufficient quantities? Creatine phosphokinase is needed by the muscle cell so that the mitochondria can create energy. Energy in a muscle cell is created by mitochondria.
Muscle weakness in Erb-Roth muscular dystrophy creates insufficient amounts of creatine phosphokinase within the muscle cell.
Types of limb-girdle muscular dystrophy
Below is a list of types of PCMD.
Type 1 dominant inheritance , i.e. only one mutation is required to manifest the disease.
Type 2 recessive inheritance , i.e. two mutations in the gene are required, one from each parent.
Some types of PCMD are given names instead of numbers.
Types of PCMD by individual names:
- Bethlem myopathy (mutation in the collagen 6 gene, dominant)
- calpainopathy (mutation in the calpain gene, recessive, another name is LGMD2A)
- dysferlinopathy (mutation in the dysferlin gene, recessive, another name is LGMD2B)
- myofibrillar myopathy (mutations in the desmin, alpha-B crystallin, myotilin, ZASP, filamin C, BAG3 or SEPN1 genes; all dominant except the desmin type, which can be either dominant or recessive)
- sarcoglycanopathy (mutation in the sarcoglycan gene; recessive; other names - LGMD2C, LGMD2D, LGMD2E, LGMD2F)
- ZASP-related myopathies (mutation in the ZASP gene; dominant; is a form of myofibrillar myopathy)
Dominant types of PCMD by numbers:
- LGMD1A / PKMD1A (mutation in the myotilin gene)
- LGMD1B / PKMD1B (mutation in the lamin A/C gene)
- LGMD1C / PKMD1C (mutation in the caveolin gene)
- LGMD1D / PKMD1D (mutation in the DNAJB6 gene)
- LGMD1E / PCMD1E , also called desmin myopathy - a type of myofibrillar myopathy (mutation in the desmin gene)
- LGMD1F / PKMD1F (mutation on chromosome 7)
- LGMD1G / PKMD1G (mutation on chromosome 4)
- LGMD1H / PKMD1H (mutation on chromosome 3)
Recessive types of PCMD by numbers:
- LGMD2A / PKMD2A (mutation in the calpain gene)
- LGMD2B / PCMD2B (mutation in the dysferlin gene)
- LGMD2C/PCMD2C , also called SCARMD1 (gamma sarcoglycan gene mutation)
- LGMD2D/PCMD2D , also called SCARMD2 (alpha sarcoglycan gene mutation)
- LGMD2E / PKMD2E (mutation in the beta sarcoglycan gene)
- LGMD2F / PKMD2F (mutation in the delta sarcoglycan gene)
- LGMD2G / PKMD2G (mutation in the telethonin gene)
- LGMD2H / PKMD2H (mutation in the TRIM32 gene)
- LGMD2I / PKMD2I (mutation in the FKRP gene)
- LGMD2J / PKMD2J (mutation in the titin gene)
- LGMD2K / PKMD2K (mutation in the POMT1 gene)
- LGMD2L / PKMD2L (mutation in the ANO5 gene)
- LGMD2M / PKMD2M (mutation in the fukutin gene)
- LGMD2N / PKMD2N (mutation in the POMT2 gene)
- LGMD2O / PKMD2O (mutation in the POMGnT1 gene)
- LGMD2Q / PKMD2Q (mutation in the plectin gene)
Source: Muscular Dystrophy Association
What does a muscle cell do to stop the release of creatine phosphokinase?
To preserve the necessary substances, the cell is forced to close these holes. And they can only be closed with substances that are larger than these holes. The muscle cell begins to retain fat components inside itself that are larger than these holes.
To keep the fatty components near these holes, you need to apply force. To exert force requires energy. Energy is created by mitochondria. Therefore, to save the life of the cell and their lives, mitochondria move from the motor proteins actin and myosin to the walls, floor and ceiling of the cell. Actin and myosin are left without some of the energy. The cell, frightened that other holes may form in the frame, is concerned about creating additional fatty inclusions inside itself (just in case). There are so many of these inclusions that the fat begins to squeeze the motor proteins of the cell until they are completely immobilized.
Rice. 2. State of the muscle cell in Erb-Roth muscular dystrophy
Explanation for the photo:
A - change in the size of muscle fibers and necrotic myofibrils (arrows).
B — cluster of basophilic regenerating myofibrils (arrows).
C , Immunohistochemical staining for dystrophin demonstrates marked loss of normal plasma membrane staining.
How does my method work to restore normal muscle movement?
Using my method, Nikolay Nikonov, for Erb-Roth dystrophy: stretching the muscle, fixing it in a certain position and pressing on it with a certain technical technique, the motor proteins are released from the pressure of fatty inclusions.
Using an electron microscope, I see an increase in the presence of fat cells and a concentration of mitochondria near the cell walls, which confirms my logical reasoning.
Rice. 3. Muscle biopsy for the diagnosis of Erb-Roth muscular dystrophy
Muscle biopsy shows severe fibrosis of the endomysial fibis ( A ) and lymphocyte infiltration ( B ). ( C ) Basophilic regenerating fibers are shown. ( D ) Necrotic myofib infiltrated with marked lymphocytes and histiocytes
Until the fatty inclusions, with their heaviness, stop the work of motor proteins, there is muscle movement. They may be weak, they may tire quickly, but there are muscle movements.
As soon as fatty inclusions clamp motor proteins, immobilization occurs. If immobilization occurs in the diaphragm, breathing stops. If immobilization occurs in the heart, then the heart stops.
I cannot restore the functioning of the gene, but... by influencing the muscles with my method, it was possible to reduce the amount of fatty components inside the muscle cell in a girl with progressive Erb-Roth muscular dystrophy and some of the mitochondria returned to their places.
The muscles began to contract and made it possible for Nastya to begin to move.
Now Nastya is at home, her condition has not worsened. There is even some improvement - Nastya started swimming in the pool.
Doctor Nikonov
The disadvantage of restoring Erb-Roth muscular dystrophy is that you need to constantly periodically influence the muscles with my method so that there are no excessively large fat deposits in the muscle cells.
With diabetes, people experience inconvenience all their lives when taking insulin, but despite this they live a full life (have a family and children).
I am proud of my knowledge, experience and skills!
The result of my knowledge on restoring normal muscle function in Duchenne muscular dystrophy:
Emine, Sergei, and Jacob gained muscle strength. They and my other patients walk like ordinary healthy people!
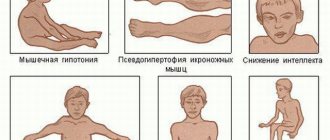
Symptoms of Erb-Roth dystrophy
Here are the key symptoms for this diagnosis, which begins to develop in children and adolescents:
- There is a delay in the child starting to walk independently.
- An uncomfortable gait for the patient, which looks like hobbling from foot to foot. This is also called the “duck” type of walking. This occurs due to a symmetrical weakening of the muscles in the area of pain.
- The child often stumbles when moving and falls when running, in other words – imbalance and instability.
- Difficulties that arise when trying to get out of bed or chair. Difficulties also occur when walking on slopes, climbing, and even going down stairs.
- There is a convexity of the scapular bones. This occurs due to weakening of the patient’s serratus anterior muscles and the rhomboid muscles of the back.
- Waist circumference decreases. This is due to the fact that with Erb-Roth muscular dystrophy, there is a decrease in the tone of the transverse muscles of the chest, abdomen and ileal rib.
- Pathological fatigue in a child.
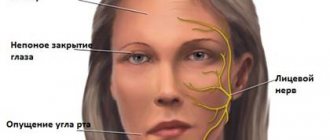
As the disease progresses, there is constant general weakness and weakening of the muscular corset of the back and shoulder girdle muscles. These processes lead to such postural defects as hyperlordosis. For patients with dystrophy, it becomes more and more difficult each time, as well as holding objects in their hands and raising their hands to the top. As for the facial muscles, they also lose their mobility. This results in incomplete closure of the eyelids and protrusion of the lips.
The process of gradual decrease in muscle tone leads to inevitable thinning and flabbiness of the patient’s muscle tissue with progressive Erb-Roth dystrophy, replacing it with adipose and fibrous tissue, i.e. myodystrophy.
Symptoms of the disease in later stages
Severe loss of muscle mass, flexion contour, contraction of the patient's tendons and almost complete loss of deep tendon reflexes in the child's lower extremities (knee and sole).
Diagnosis of Erb-Roth muscular dystrophy
- The diagnosis of the disease in question is based on a physical examination of patients, a study of the patient’s family history and subsequent analysis of the collected data.
- Genetic testing is carried out. Necessary for subsequent accurate determination of muscular dystrophy.
- Electroneuromyography.
- A biopsy of muscle tissue is performed with biochemical testing.
- A general blood test is given.
- A blood test for creatine phosphokinase is taken.
- Analysis of the patient's urine.
As for electromyography, it makes it possible to study not only the degree of neuromuscular transmission, but also to determine the level of direct muscle excitability, which is extremely important for the differential diagnosis of the disease with pathologies of neuropathic muscles.
What is limb-girdle muscular dystrophy?
Translator's note.
The name varies in different sources: limb-girdle MD and limb-girdle MD. Limb-girdle muscular dystrophy (LGMD) is not one common disease. This is a whole group of diseases that affect muscles, mainly located in the hips and shoulders.
The shoulder girdle is the bony structure that surrounds the shoulder region.
(Information from Wikipedia: the shoulder girdle (girdle of the upper limbs) is a set of bones (pairs of shoulder blades and collarbones) and muscles that provide support and movement of the upper (front) limbs.)
The pelvic girdle is a bony structure surrounding the hip area.
Collectively they are called “limb belts”. In PCMD, the muscles that are most damaged are those connected to the bones of these girdles.
The term " proximal " is also used to describe the muscles damaged in PCMD. Proximal muscles are muscles located close to the center of the body. Distal muscles are muscles that are further away from the center of the body (for example, muscles of the hands or feet). Distal muscles in PCMD are affected at later stages, but may retain their function.
As of the end of 2012, there are more than 20 different subtypes of PCMD. This is a complex and constantly evolving area of research.
Treatment of Erb-Roth dystrophy
It should be noted right away that I cannot restore the damaged gene, but...
Doctor Nikonov
The impact of my method is aimed at reducing the intensity of symptoms, slowing down the progression of the disease, increasing muscle strength, and restoring proper movement in all muscle groups.
I do not treat Erb-Roth muscular dystrophy. I am working on restoring normal muscle function . Therefore, I will not describe the treatment procedures used in hospitals and other rehabilitation centers in my article. Chances are you've tried them.
My article is for informational purposes only. I took the information from my observations and ten years of research and discoveries of scientists from all over the world.
Medical certificate
The disease is a polymorphic variant of hereditary myodystrophy. It differs from other types of pathology in its clinical picture, course and time of onset. The description of the disease was first presented by the German neurologist W. Erb in 1882. At the same time, V. Roth was working on this problem in Russia, which he later designated as “tabes muscularis.” It was after the names of the two scientists that the disease was named. In modern neurology, several of its names are used - progressive Erb-Roth muscular dystrophy, limb-girdle muscular dystrophy.
Pathology begins its development, as a rule, in childhood. However, the age at which symptoms first appear can range from 10 to 30 years. Men and women suffer equally from the manifestations of muscular dystrophy. Neurologists note that the disease, which began in childhood, progresses rapidly when compared with its course in adolescence and adulthood. In addition, in the second case it occurs in a mild form.