Причины
Заболевание наследственное, сцепленное с X-хромосомой, поэтому болеют практически всегда мальчики. Девочки являются носителем патологического гена (мальчики редко доживают до половозрелого возраста, к тому же, как правило, стерильны). В хромосоме происходит изменение структуры гена, отвечающего за синтез белка дистрофина.
Хоть содержание дистрофина в скелетной мускулатуре предельно мало (тысячные доли процента), без него быстро развивается некроз мышечной ткани, развивается прогрессирующая дистрофия мышц. Если ген повреждается на участке, полностью разрушающем синтез белка-дистрофина, развивается дистрофия Дюшенна. При вовлечении в процесс малозначимых отделов белка, заболевание принимает форму дистрофии Беккера.
Общее описание врождённых мышечных дистрофий
Перевод материалов Muscular Dystrophy Association. Ссылка на источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Что такое врождённая мышечная дистрофия (ВМД, англ. congenital muscular dystrophy (CMD)?
Врождённая мышечная дистрофия относится к группе мышечных дистрофий, которые становятся заметными при рождении или сразу после рождения.
Мышечные дистрофии являются, в основном, генетическими дегенеративными заболеваниями, поражающие большей частью произвольно сокращающиеся мышцы.
Дети с врождённой мышечной дистрофией — слабые уже при рождении и могут испытывать трудности с дыханием и глотанием.
В настоящее время методы поддержки больных с ВМД значительно улучшились и повысили выживаемость пациентов, а клинические испытания препаратов против этой болезни — уже не в таком далёком будущем, как это было раньше.
Более подробно см. на mda.org: Types of CMD.
Симптомы ВМД
ВМД приводит к общей мышечной слабости с возможным развитием суставных контрактур, также возможно ослабление суставов.
В зависимости от типа при ВМД может появиться искривление позвоночника, дыхательная недостаточность, ментальные нарушения, сложности с обучением, дефекты глаз или судороги.
Более подробно см. на mda.org: Types of CMD and Signs and Symptoms.
Причины появления ВМД
ВМД возникает из-за мутаций в генах, отвечающих за определённые белки, необходимые для мышц, а в некоторых случаях — для глаз или мозга.
Более подробно см. на mda.org: Causes/Inheritance.
Прогрессирование ВМД
ВМД проявляется при рождении или через короткое время после него. Прогрессирование заболевание зависит от типа. Многие типы прогрессируют медленно, но некоторые приводят к сокращению продолжительности жизни.
Статус исследований по ВМД
Исследователи определили многие из генов, дефекты в которых приводят к появлению ВМД. Эти открытия ведут нас к лучшему пониманию природы болезни и улучшают диагностику, а также стратегии лечения.
Более подробно см. на mda.org: Research.
Источник: https://www.mda.org/disease/congenital-muscular-dystrophy
Симптомы
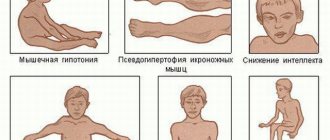
Начало развития симптомов начинается в раннем детском возрасте, чаще от 1 до 3 лет. Изначально отмечается отставание в моторном развитии, ребенок поздно начинает ходить, часто спотыкается при ходьбе, быстро устает. Позже развивается постоянная патологическая утомляемость мышц. Ребенок практически не может взбираться по лестнице. Походка начинает напоминать «утиную».
Характерным симптомом является симптом «лесенки»: попытка встать из положения сидя происходит с использованием рук, постепенно, медленно, в несколько этапов.
Постепенно начинает отмечаться атрофия мышц, вначале проксимальных отделов нижних, потом верхних конечностей. Позже атрофируются мышцы тазового пояса, бедер, спины, плечевого пояса. Почти всегда развивается «осиная» талия, искривление позвоночника, выпирание лопаток (крыловидные лопатки).
Практически всегда имеет место характерный симптом прогрессирующей мышечной дистрофии Дюшенна – псевдогипертрофия мышц голеней. Мышцы, хоть и увеличены в объеме, они не имеют достаточной силы, очень болезненны на ощупь.
Можно выделить три стадии заболевания: — I стадия – слабость проявляется лишь при значимой физической нагрузке (обычно первый год течения болезни). — II стадия – затруднен подъем по лестнице, быстро развивается слабость при ходьбе. — III стадия – представляет собой параличи, контрактуры мышц с невозможностью cсамостоятельного передвижения.
По видам течения подразделяется на: Быстрое прогрессирование. Способность к передвижению утрачивается быстро, в течение первых 4-5 лет с начала заболевания. Средний темп прогрессирования: передвигаться пациент не может спустя 10 лет. Медленное прогрессирование: выраженных двигательных нарушений через 10 лет от начала болезни нет. Обычно такой вариант характерен для других типов мышечных дистрофий, нежели дистрофии Дюшенна.
Прогрессирующие мышечные дистрофии
Прогрессирующие мышечные дистрофии (ПМД) — гетерогенная группа наследственных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофией скелетных мышц.
КЛИНИЧЕСКАЯ КАРТИНА
Для всех ПМД типичны мышечная слабость различной степени выраженности и мышечные атрофии. Тип распределения мышечной слабости при ПМД — один из основных диагностических критериев. Для каждой из форм ПМД характерно избирательное поражение определённых мышц при сохранности других, рядом расположенных. В целом типичный миопатический симптомокомплекс включает следующие признаки. • Симметричную проксимальную мышечную слабость различной степени выраженности (мышечная сила от 3-4 баллов на ранней и до 1-0 — на поздних стадиях заболевания) , постепенно развивающиеся атрофии мышц. • Симптом Говерса: больной, для того чтобы подняться из положения на корточках, опирается руками об пол, затем поднимается, опираясь руками об колени, — «взбирается по себе». Этот рано появляющийся симптом обусловлен слабостью мышц бёдер и тазового пояса. • Затруднения при ходьбе по лестнице — больной помогает себе с помощью рук. • «Утиную» (переваливающуюся) походку, связанную со слабостью мышц тазового пояса. • Поясничный гиперлордоз, обусловленный слабостью мышц тазового пояса и спины. • «Крыловидные» лопатки вследствие слабости передней зубчатой мышцы, а также других мышц, фиксирующих лопатку. • Псевдогипертрофию икроножных мышц вследствие развития в них соединительной ткани (сила мышц при этом снижена) . • Ходьбу на цыпочках из-за контрактур ахилловых сухожилий. • Сохранность экстраокулярных мышц, мышц лица. Миопатический симптомокоплекс наиболее отчётливо выявляют при ПМД Дюшенна и Беккера. • Для ПМД Дюшенна характерно раннее начало заболевания (в 3-7 лет) , быстрое прогрессирование, высокие показатели КФК, выраженная спонтанная активность по данным игольчатой ЭМГ, отсутствие дистрофина в мышцах при иммуногистохимическом исследовании. По мере прогрессирования мышечной слабости затрудняется самостоятельная ходьба и уже в 9-15 лет больные вынуждены пользоваться инвалидным креслом, что провоцирует развитие кифосколиоза, остеопороза. На поздних стадиях у большинства больных развиваются дилатационная кардиомиопатия, слабость дыхательной мускулатуры. Интеллект чаще всего умеренно снижен. • Клинические про явления ПМД Беккера в целом напоминают таковые при форме Дюшенна, но течение заболевания более мягкое: дебют приходится на более поздний возраст (от 2 до 21 года, в среднем в 11 лет) , летальный исход наступает позже (в 23-63 года) . • Конечностно-поясные формы ПМД также характеризуются развитием миопатического симптомокомплекса. ПМД Эрба по возрасту начала заболевания, скорости прогрессирования и клиническим проявлениям напоминает форму Беккера, однако для неё не характерна кардиальная патология, кроме того, заболевание отмечают как у мальчиков, так и девочек. При других конечностнопоясных формах возможны слабость мышц лица и кардиомиопатия. • ПМД Ландузи-Дежерина характеризуется выраженной слабостью мимических мышц (за исключением редкой формы без мимической слабости), симптомом «крыловидных» лопаток, слабостью дву- и трёхглавых мышц плеча при интактных дельтовидных мышцах, степпажем. Как правило, интактными остаются экстраокулярные мышцы (за исключением одного подтипа) и мышцы языка и глотки, дыхательная мускулатура. У некоторых больных возникает слабость мышц тазового пояса (около 20% больных вынуждены пользоваться инвалидным креслом) . Мышечные атрофии часто бывают асимметричными. У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма. • ПМД Эмери-Дрейфуса характеризуется наличием контрактур (чаще в локтевых, коленных суставах, задних мышцах шеи, из-за которых голова оказывается слегка запрокинутой) и плечелопаточно-перонеальным распределением мышечной слабости и атрофий с сохранностью лицевой мускулатуры. Часто отмечают нарушения ритма сердца и кардиомиопатию. Заболевание часто дебютирует с контрактур. • Основной симптом офтальмофарингеальной формы — хроническая прогрессирующая наружная офтальмоплегия, затем присоединяется умеренный бульбарный синдром. В дальнейшем развивается проксимальная мышечная слабость в руках и ногах. • Дистальные миопатии характеризуются преобладанием слабости дистальных мышц. При миопатии Веландер в наибольшей степени поражаются разгибатели кистей, при миопатии Миоши — икроножные мышцы: больные плохо стоят на носках, часто спотыкаются. При миопатии Говерса, тибиальной миопатии главный симптом — степпаж из-за слабости перонеальной группы мышц, при этом миопатия Говерса склонна к дальнейшей генерализации: через 5-10 лет присоединяется слабость кистей и мышц шеи, часто о 1 пальца на ногах и V — на руках. При тибиальной миопатии, распространённой в Финляндии, чаще всего наблюдают изолированное поражение передних больше берцовых мышц, иногда развивается кардиомиопатия.
СИМПТОМЫ
При ПМД Дюшенна, Беккера, конечностно-поясных формах проявляется наиболее выраженная слабость в пояснично-подвздошных мышцах, мышцах бёдер, дельтовидных, дву- и трёхглавых мышцах плеча. Менее выражена слабость в дистальных мышцах конечностей. Лицевые мышцы остаются сохранными. Наряду с мышечной слабостью постепенно развиваются гипотрофии поражённых мышц вплоть до атрофии на поздних стадиях. При этом соседние мышцы могут быть полностью клинически интактны.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов. Немедикаментозное лечение Чрезмерная физическая нагрузка, как и недостаточная, приводит к нарастанию мышечной слабости. Ежедневная ЛФК позволяет поддерживать мышечный тонус и препятствует развитию контрактур. Комплекс ЛФК обязательно должен включать активные и пассивные упражнения, упражнения на растяжку/предупреждение контрактур и дыхательную гимнастику. Активный массаж с разминанием мышц может усиливать мышечную слабость и утомляемость, поэтому рекомендуют щадящий массаж. Физиотерапевтическое лечение больные переносят по-разному: некоторые не ощущают улучшений или даже жалуются на усиление мышечной слабости. Хирургическое лечение В некоторых случаях возможно хирургическое лечение контрактур, однако при этом необходимо помнить о возможности увеличения мышечной слабости за время восстановительного лечения (вплоть до потери способности к ходьбе). В ряде случаев необходима имплантация кардиостимулятора.
Диагностика
Клиническая картина очень яркая. Часто заболевание ставится после выяснения генетического анамнеза (наличие случаев в семье), неврологического осмотра. В неврологическом статусе отмечается пропадание коленных рефлексов, чуть позже исчезают рефлексы с бицепса, трицепса. Ахилловы рефлексы долгое время сохранны.
Внешне может выявиться деформация суставов стопы, имеются признаки кардиомиопатии: нарушение пульса, глухость сердечных тонов, расширение полостей сердца по ЭхоКГ, изменения на электрокардиограмме.
Важным фактором является повышение биохимических показателей креатинфосфокиназы (фермент-показатель распада мышц). Активность данного фермента увеличивается в десятки раз. Имеется прямая корреляция между степенью увеличения активности фермента и выраженностью проявлений дистрофии Дюшенна. [!] В сложных диагностических ситуациях проводят цитологическое исследование.
Лечение и прогноз жизни
Лечение симптоматическое. Используются гормональные препараты для остановки разрушения мышечного волокна, фосфолипиды в качестве защиты клеток мышц от разрушения, элементы лечебной гимнастики. Внедряются в практику различные ортопедические приспособления для облегчения передвижения. Массаж строго противопоказан в большинстве случаев, так как может приводить к ускорению распада мышц. Лечение наследственных заболеваний – дело будущего.
Прогноз жизни для пациентов неблагоприятный. Течение заболевание прогрессирующее. Неизбежен летальный исход. Как правило, к семилетнему возрасту развивается выраженная симптоматика, приводящая к 13-14 годам к полной обездвиженности. Больные редко доживают до 18-20 лет.