IIMs are included in the group of autoimmune inflammatory pathologies of neuromuscular tissue, which are observed as independent nosological forms, or as syndromes in the presence of various types of autoimmune diseases, mainly of a rheumatic nature. The pathology is characterized by non-purulent inflammation of the muscles called skeletal muscles. Such pathologies include: polymyositis (PM), and also (in case of damage to the skin) – dermatomyositis (DM).
The true prevalence of the disease is not precisely known. In some cases, it has a poor prognosis. Differential diagnosis is carried out with great difficulties.
Known types and forms of myositis:
- in combination with CTD (crossover syndrome);
- ossifying;
- localized or focal;
- with the presence of intracellular inclusions;
- giant cell;
- eosinophilic;
- in combination with tumors;
- polymyositis (PM);
- dermatomyositis (DM);
- juvenile DM;
As can be seen from the statistics, 60% of autoimmune pathologies have the following neurological disorders:
- polyneuropathies in various variants (Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, multifocal motor neuropathy);
- synaptic diseases associated with disorders of neuromuscular transmission (myasthenia gravis, Lambert-Eaton myasthenic syndrome);
- IVM (PM, DM, and so-called inclusion body myositis);
- a number of pathologies of the central nervous system (for example, multiple sclerosis and rigid person syndrome).
So, IIM is a pronounced disease of an autoimmune nature and, at the same time, one of the neuromuscular pathologies. Historically, dermatomyositis and polymyositis have been studied for about 150 years, so by now there is enough clinical and pathomorphological material in medicine. Some forms of the course of IVM are similar to already studied variants of myopathies, polyneuritis, myasthenia, hence such medical terms as “pseudomyopathic”, “pseudomyasthenic”, “pseudopolyneuritic” and other types of PM.
Classification options for idiopathic inflammatory myopathies
Unfortunately, there is no unified classification of IVM yet. Bohan and Peter back in 1975. The following forms have been proposed:
- primary (idiopathic) polymyositis;
- primary (idiopathic) dermatomyositis;
- PM (DM) in combination with a neoplastic process;
- PM (PM) in combination with other diffuse connective tissue diseases;
- juvenile polymyositis.
No new criteria for the classification of IIM have yet been presented, in addition to the proposal to identify certain myositis-specific antibodies during diagnosis. The classification that was developed by I. Gusmanova-Petrusevich is closest to the tasks of clinical diagnostics.
It distinguishes the following types:
- DM and PM (as a separate form of ICM);
- inflammatory diseases of muscle tissue (myositis) in the presence of primary severe collagenosis (scleroderma, periarteritis nodosa, systemic lupus erythematosus);
- granulomatous PM;
- various types of parasitic myositis;
- polymyalgia of a rheumatic nature.
According to the International Classification of Diseases, 10th revision (ICD-10), IVM and IVM with the presence of cross-connective tissue diseases are divided into the following types:
- IVM
- DM (M33.1 according to ICD-10);
- PM (M33.2 according to ICD-10);
- myositis with inclusions (M33.1 according to ICD-10);
- dermatopolymyositis, unspecified (M33.9 according to ICD-10);
- dermatopolymyositis in neoplasms C00-D48 (M36.0* according to ICD-10).
- Inflammatory myopathies in combination with connective tissue pathologies
- systemic lupus erythematosus (M32 according to ICD-10);
- scleroderma (M34 according to ICD-10);
- rheumatoid arthritis (MO5.3 according to ICD-10);
- other connective tissue diseases (M35 according to ICD-10).
Diagnostic methods
Diagnosis of IVM is carried out based on the following indicators:
- clinical picture with the presence of a characteristic pattern of movement disorders;
- laboratory tests - ALT, AST, lactate dehydrogenase, aldolase and CPK (cretinin phosphokinase) a marker of muscle necrosis. With massive muscle damage, a decrease in serum creatinine levels may be observed;
- immunological tests - ANA, myositis-specific and myositis-associated antibodies;
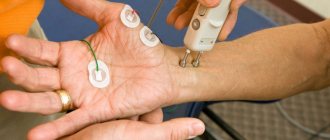
- electroneuromyographic examination (EMG) (primarily using needle electrodes);
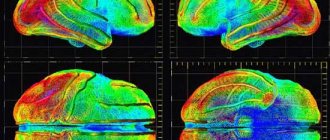
- magnetic resonance imaging (MRI), which identifies areas of inflammation and edema during active myositis and fibrosis;
- magnetic resonance spectroscopy (MRS), which detects a decrease in muscle metabolism in affected muscles and is used as one of the non-invasive research methods;
- Ultrasound is used both to identify myositis and for differential diagnosis, for example, lumbar-limb muscular dystrophies, amyotrophic lateral sclerosis.
- The main symptom of polymyositis and dermatomyositis is a fairly rapidly developing weakness in the muscles, in which, as a rule, the proximal muscles of the arms and legs are affected (tendon reflexes are preserved. Neither with PM nor with DM is there any specific clinical picture. Therefore, quite For a long time they were considered to be one and the same disease, but now doctors have established that these are two different diseases, despite the similarity of some symptoms.
Symptoms of progressive muscular dystrophies
In most cases, pathologies begin to manifest themselves as slight muscle weakness in the limbs, faster fatigue after walking and physical activity. Over the years, weakness increases, progressive muscle atrophy develops, and limbs become deformed.
Pathological changes are symmetrical for the muscles of the trunk and limbs. Muscle atrophy is most often observed in the proximal limbs. As muscle weakness increases, tendon reflexes gradually fade away and muscle tone decreases. Peripheral flaccid paralysis develops, joint contractures occur.
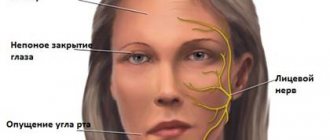
Progressive muscular dystrophies can affect facial muscles and provoke dysarthia. They can affect the respiratory muscles, causing congestive pneumonia and respiratory failure. The consequences of such pathologies also include cardiomyopathy, heart failure, dysphagia, and myopathic laryngeal paresis.
Features of some forms of the disease
Juvenile Erb myopathy is characterized by autosomal recessive inheritance. Appears at 20–30 years of age. The pathology affects the muscles of the thighs and pelvic floor, then spreads to other areas of the body.
Pseudohypertrophic Duchenne myopathy is characterized by sex-linked (boys are more often affected) recessive inheritance. The disease usually manifests itself at the age of 3 years. It begins with atrophy of the muscles of the pelvic girdle and proximal parts of the lower extremities, accompanied by hypertrophy of the calf muscles.
Landouzy-Dejerine myopathy is a pathology with autosomal dominant inheritance, manifested by damage to the facial muscles at 10–20 years of age. Gradually, the muscles of the chest, shoulder girdle, and shoulders are involved in the process.
Scapuloperoneal myopathy is a disease of autosomal dominant inheritance. Atrophy develops in the distal legs and proximal arms. Mild sensory disturbances of the distal upper and lower extremities may occur.
Oculopharyngeal myopathy is accompanied by damage to the extraocular muscles, weakness of the muscles of the pharynx and tongue. The manifestation of pathology occurs at the age of 4–6 years.
Distal late myopathy is characterized by autosomal dominant inheritance. Accompanied by weakness and atrophy of the distal limbs.
Therapy of idiopathic inflammatory myopathies in our clinic
Treatment of polymyositis/dermatomyositis is a major problem due to the rarity and heterogeneity of IIM categories, and has two main directions: restoration of muscle strength and relief of extramuscular manifestations.
The difficulty in therapy is dictated by the development of fibrous and fatty involution of muscle tissue. The main thing in treatment measures is to achieve a complete clinical response or remission of the disease.
Basic principles of treatment:
- glucocorticoids (glucocorticosteroids);
- basic anti-inflammatory drugs.
The basis of therapy to control the activity of the pathology remains glucocorticoids (glucocorticosteroids), prescribed in an amount of 1 – 1.5 mg/kg per day. It must be emphasized that, unlike the result in other rheumatic diseases, the effect of this therapeutic method is achieved slowly. It takes several months to restore muscle strength and normalize CPK (depending on the initial severity of the disease at the time of treatment). The maximum dose of GC is used for 2–3 months (in the absence of compelling reasons to reduce it). Further management of the patient is individual and depends on his condition during dynamic observation (monitoring muscle strength, creatine phosphokinase levels, electromyography parameters). As a rule, the rate of reduction in glucocorticoid intake is ½, then ¼ tablet, over 7 to 10 days, to an individually selected maintenance dose. If there is no “response” to GC, it is necessary to reconsider the diagnosis, and if it is confirmed, increase the dosage. Most patients who are resistant to corticosteroids are most likely to have inclusion body myositis, necrotizing myopathy, or progressive muscular dystrophy.
Second-line drugs include drugs such as azathioprine, methotrexate, cyclophosphamide, mycophenolate mofetil and hydroxychloroquine. A special place is occupied by intravenous immunoglobulin (IVIG). Recent studies have confirmed its effectiveness (when taken in high doses in combination with GC for several years) in resistant types of polymyositis/dermatomyositis, severe forms of dysphagia and IPL. Positive dynamics are noted after the introduction of three monthly IVIG injections in patients with an early stage of the disease, as well as resolution of fibrotic changes and stable remission for 2 years, without the use of other immunosuppressants. Some hope is pinned on the use of low-dose IVIT for inclusion body myositis.
It is also advisable to use plasmapheresis, mainly in patients with severe MVM, resistant to other treatment methods, in combination with GC and MT.
Rehabilitation measures aimed at optimizing mobility, preventing the formation of contractures, as well as reducing the risk of osteoporosis and weight gain due to the administration of high doses of glucocorticosteroids and immobilization are important. Physical activity is carried out differentially depending on the stage of the disease.
After completion of IVM therapy, patients are recommended to regularly visit a rheumatologist for examination and preventive measures to prevent relapses.
Myopathies
Juvenile Erb's myopathy
inherited autosomal recessively. Pathological processes begin to appear at the age of 20-30 years. First of all, they cover the muscles of the pelvic girdle and hips, then quickly spread to other muscle groups. Involvement of the facial muscles is not typical. The onset of myopathy at a younger age leads to early immobility of patients. When the disease develops at an older age, its course is less severe: patients retain the ability to move for a long time.
Pseudohypertrophic Duchenne myopathy
inherited in a sex-linked recessive manner. Only boys are affected. As a rule, it manifests itself during the first 3 years of life, less often - in the period from 5 to 10 years. Typically, it begins with atrophic changes in the muscles of the pelvic girdle and proximal legs, accompanied by pseudohypertrophy of the calf muscles. Contractures and curvature of the spine (kyphosis, scoliosis, hyperlordosis) occur early. Oligophrenia may occur. The disease occurs with damage to the respiratory muscles and heart (cardiomyopathy is observed in 90% of patients with Duchenne myopathy), which causes early death.
Landouzi-Dejerine myopathy
has autosomal dominant inheritance. Manifests at 10-20 years of age with damage to the facial muscles. Gradually, weakness and atrophy cover the muscles of the shoulder girdle, shoulders and chest. The muscles of the pelvic girdle are usually not affected. Characterized by a slow course with long-term preservation of performance, without a reduction in life expectancy.
Scapuloperoneal myopathy
- an autosomal dominant disease. Its peculiarity is the development of atrophy in the muscles of the distal legs and proximal arms, as well as the presence of mild sensory disturbances in the distal parts of both lower and upper extremities.
Oculopharyngeal myopathy
characterized by a combination of damage to the extraocular muscles with weakness of the muscles of the tongue and pharynx. Usually manifests with bilateral ptosis, followed by swallowing disorders. A feature of this myopathy is its late onset - in the 4th-6th decade of life.
Distal late myopathy
inherited in an autosomal dominant manner. It is characterized by the development of weakness and atrophy in the distal parts of the extremities: first in the feet and hands, and then in the legs and forearms. Characterized by a slow flow.
Features of clinical manifestations of various forms of congenital, hereditary and metabolic myopathies are described in independent reviews.