Causes
The disease is hereditary, linked to the X chromosome, so boys are almost always affected. Girls are carriers of a pathological gene (boys rarely survive to adulthood, and, moreover, are usually sterile). A change in the structure of the gene responsible for the synthesis of the dystrophin protein occurs on the chromosome.
Although the content of dystrophin in skeletal muscles is extremely small (thousandths of a percent), without it necrosis of muscle tissue quickly develops and progressive muscle dystrophy develops. If the gene is damaged in a site that completely destroys the synthesis of the dystrophin protein, Duchenne dystrophy develops. When insignificant parts of the protein are involved in the process, the disease takes the form of Becker's dystrophy.
General description of congenital muscular dystrophies
Translation of materials from the Muscular Dystrophy Association. Source link: https://www.mda.org/disease/congenital-muscular-dystrophy
What is congenital muscular dystrophy (CMD)?
Congenital muscular dystrophy refers to a group of muscular dystrophies that become noticeable at birth or immediately after birth.
Muscular dystrophies are mainly genetic degenerative diseases that mostly affect voluntary muscles.
Children with congenital muscular dystrophy are weak at birth and may have difficulty breathing and swallowing.
Currently, methods of supporting patients with AMD have significantly improved and increased patient survival, and clinical trials of drugs against this disease are no longer in such a distant future as they once were.
For more information, see mda.org: Types of CMD.
Symptoms of AMD
AMD leads to general muscle weakness with the possible development of joint contractures, and weakening of the joints is also possible.
Depending on the type, AMD may cause spinal curvature, respiratory failure, mental problems, learning difficulties, eye defects, or seizures.
For more information, see mda.org: Types of CMD and Signs and Symptoms.
Causes of AMD
AMD occurs due to mutations in genes that control certain proteins needed by the muscles and, in some cases, the eyes or brain.
For more information, see mda.org: Causes/Inheritance.
Progression of AMD
AMD appears at birth or shortly thereafter. The progression of the disease depends on the type. Many types progress slowly, but some lead to a shortened lifespan.
Status of research on AMD
Researchers have identified many of the genes whose defects lead to AMD. These discoveries lead us to a better understanding of the nature of the disease and improve diagnosis as well as treatment strategies.
For more information, see mda.org: Research.
Source: https://www.mda.org/disease/congenital-muscular-dystrophy
Symptoms
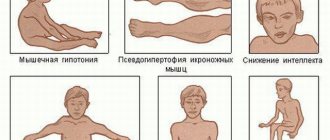
The onset of symptoms begins in early childhood, usually between 1 and 3 years. Initially, there is a delay in motor development, the child begins to walk late, often stumbles when walking, and gets tired quickly. Later, constant pathological muscle fatigue develops. The child practically cannot climb stairs. The gait begins to resemble a “duck” gait.
A characteristic symptom is the “ladder” symptom: an attempt to get up from a sitting position occurs using the hands, gradually, slowly, in several stages.
Gradually, muscle atrophy begins to be noted, first in the proximal parts of the lower, then upper extremities. Later, the muscles of the pelvic girdle, hips, back, and shoulder girdle atrophy. A “wasp” waist, curvature of the spine, and protruding shoulder blades (pterygoid blades) almost always develop.
Almost always there is a characteristic symptom of progressive Duchenne muscular dystrophy - pseudohypertrophy of the leg muscles. The muscles, although increased in volume, do not have sufficient strength and are very painful to the touch.
Three stages of the disease can be distinguished: - Stage I - weakness manifests itself only with significant physical activity (usually the first year of the disease). — Stage II – difficulty climbing stairs, weakness when walking quickly develops. — Stage III – represents paralysis, muscle contractures with the inability to move independently.
According to the type of course, it is divided into: Rapid progression. The ability to move is lost quickly, within the first 4-5 years from the onset of the disease. Average rate of progression: the patient cannot move after 10 years. Slow progression: there are no pronounced motor disorders 10 years after the onset of the disease. Typically, this option is characteristic of other types of muscular dystrophies than Duchenne dystrophy.
Progressive muscular dystrophies
Progressive muscular dystrophies (PMDs) are a heterogeneous group of inherited diseases characterized by progressive muscle weakness and skeletal muscle atrophy.
CLINICAL PICTURE
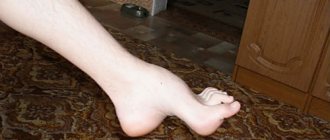
All PMD are characterized by muscle weakness of varying severity and muscle atrophy. The type of distribution of muscle weakness in PMD is one of the main diagnostic criteria. Each form of PMD is characterized by selective damage to certain muscles while sparing others nearby. In general, a typical myopathic symptom complex includes the following symptoms. • Symmetrical proximal muscle weakness of varying severity (muscle strength from 3-4 points in the early stages and up to 1-0 in the later stages of the disease), gradually developing muscle atrophy. • Govers' symptom: the patient, in order to rise from a squatting position, rests his hands on the floor, then rises, leaning his hands on his knees - “climbing on his own.” This early-onset symptom is caused by weakness of the muscles of the hips and pelvic girdle. • Difficulty walking up stairs - the patient helps himself with his hands. • “Duck” (waddling) gait associated with weakness of the pelvic girdle muscles. • Lumbar hyperlordosis, caused by weakness of the muscles of the pelvic girdle and back. • “Wing-shaped” scapula due to weakness of the serratus anterior muscle, as well as other muscles that fix the scapula. • Pseudohypertrophy of the calf muscles due to the development of connective tissue in them (muscle strength is reduced). • Walking on tiptoes due to Achilles tendon contractures. • Preservation of extraocular muscles, facial muscles. The myopathic symptom complex is most clearly identified in Duchenne and Becker PMD. • Duchenne PMD is characterized by an early onset of the disease (at 3-7 years), rapid progression, high CK levels, pronounced spontaneous activity according to needle EMG, and the absence of dystrophin in the muscles in immunohistochemical studies. As muscle weakness progresses, independent walking becomes difficult, and already at the age of 9-15, patients are forced to use a wheelchair, which provokes the development of kyphoscoliosis and osteoporosis. In the later stages, most patients develop dilated cardiomyopathy and weakness of the respiratory muscles. Intelligence is most often moderately reduced. • Clinical manifestations of Becker's PMD generally resemble those of the Duchenne form, but the course of the disease is milder: the onset occurs at a later age (from 2 to 21 years, on average 11 years), death occurs later (at 23-63 years ) . • Limb-girdle forms of PMD are also characterized by the development of a myopathic symptom complex. In terms of the age of onset of the disease, rate of progression and clinical manifestations, Erb's PMD resembles the Becker form, however, it is not characterized by cardiac pathology, in addition, the disease is noted in both boys and girls. In other limb-girdle forms, weakness of the facial muscles and cardiomyopathy are possible. • Landouzy-Dejerine PMD is characterized by severe weakness of the facial muscles (with the exception of a rare form without facial weakness), the symptom of “pterygoid” scapulae, weakness of the biceps and triceps brachii muscles with intact deltoid muscles, stepping. As a rule, the extraocular muscles (with the exception of one subtype) and the muscles of the tongue and pharynx, and the respiratory muscles remain intact. Some patients experience weakness of the pelvic girdle muscles (about 20% of patients are forced to use a wheelchair). Muscle atrophy is often asymmetrical. Many patients experience hearing loss, cardiomyopathy, or heart rhythm disturbances. • Emery-Dreyfus PMD is characterized by the presence of contractures (usually in the elbow, knee joints, and posterior neck muscles, due to which the head is slightly thrown back) and a scapulohumeral-peroneal distribution of muscle weakness and atrophy with preservation of the facial muscles. Cardiac arrhythmias and cardiomyopathy are common. The disease often debuts with contractures. • The main symptom of the ophthalmopharyngeal form is chronic progressive external ophthalmoplegia, followed by moderate bulbar syndrome. Subsequently, proximal muscle weakness develops in the arms and legs. • Distal myopathies are characterized by predominant weakness of the distal muscles. With Welander myopathy, the extensors of the hands are most affected; with Mioshi myopathy, the calf muscles are most affected: patients stand poorly on their toes and often stumble. With Govers' myopathy, tibial myopathy, the main symptom is stepping due to weakness of the peroneal muscle group, while Govers' myopathy is prone to further generalization: after 5-10 years, weakness of the hands and neck muscles is added, often about 1 toe and V on the arms . With tibial myopathy, which is common in Finland, isolated damage to the anterior tibial muscles is most often observed, and sometimes cardiomyopathy develops.
SYMPTOMS
With PMD Duchenne, Becker, limb-girdle forms, the most pronounced weakness is manifested in the lumbar-iliac muscles, muscles of the hips, deltoid, biceps and triceps muscles of the shoulder. Weakness in the distal muscles of the extremities is less pronounced. The facial muscles remain intact. Along with muscle weakness, hypotrophy of the affected muscles gradually develops, up to atrophy in the later stages. In this case, neighboring muscles can be completely clinically intact.
TREATMENT
Treatment should be carried out exclusively by a neurologist. Self-medication is unacceptable. There is currently no definitive treatment for PMD. The goal of treatment is to maintain muscle strength, prevent the development of contractures and joint deformities. Non-drug treatment Excessive physical activity, as well as insufficient physical activity, leads to an increase in muscle weakness. Daily exercise therapy helps maintain muscle tone and prevents the development of contractures. A physical therapy complex must include active and passive exercises, stretching exercises/contracture prevention exercises and breathing exercises. Active massage with muscle kneading can increase muscle weakness and fatigue, so a gentle massage is recommended. Patients tolerate physiotherapeutic treatment differently: some do not feel any improvement or even complain of increased muscle weakness. Surgical treatment In some cases, surgical treatment of contractures is possible, but it is necessary to remember the possibility of increasing muscle weakness during rehabilitation treatment (up to loss of the ability to walk). In some cases, implantation of a pacemaker is necessary.
Diagnostics
The clinical picture is very bright. Often the disease is diagnosed after clarifying a genetic history (presence of cases in the family) and a neurological examination. In the neurological status, the disappearance of knee reflexes is noted, a little later reflexes from the biceps and triceps disappear. Achilles reflexes are preserved for a long time.
Externally, deformation of the joints of the foot may be revealed, there are signs of cardiomyopathy: irregular pulse, dullness of heart sounds, dilation of the heart cavities according to EchoCG, changes in the electrocardiogram.
An important factor is the increase in the biochemical parameters of creatine phosphokinase (an enzyme indicator of muscle breakdown). The activity of this enzyme increases tens of times. There is a direct correlation between the degree of increase in enzyme activity and the severity of manifestations of Duchenne dystrophy. [!] In difficult diagnostic situations, a cytological examination is performed.
Treatment and life prognosis
Treatment is symptomatic. Hormonal drugs are used to stop the destruction of muscle fiber, phospholipids to protect muscle cells from destruction, and elements of therapeutic exercises. Various orthopedic devices are being introduced into practice to facilitate movement. Massage is strictly contraindicated in most cases, as it can lead to accelerated muscle breakdown. Treatment of hereditary diseases is a matter of the future.
The life prognosis for patients is unfavorable. The course of the disease is progressive. Death is inevitable. As a rule, by the age of seven, severe symptoms develop, leading to complete immobility by the age of 13-14. Patients rarely live to 18-20 years of age.