Causes
Aicardi syndrome probably results from a new mutation in a gene located on the X chromosome. The exact gene or genetic mechanism that causes Aicardi syndrome is not yet known. A recently discovered report describing changes in the TEAD1 and OCEL1 genes in two girls with the syndrome was not confirmed in a large group of other girls with Aicardi syndrome. Thus, these genes do not appear to be the cause of Aicardi syndrome. The condition is believed to be fatal to newborn male children.
The parents of a woman with Aicardi syndrome are usually not affected. There have been no reports of transmission of Aicardi syndrome from the affected mother to her child. Other family members are also usually not at increased risk.
Genetics
Aicardi syndrome is a sex-linked disease; the defective gene is located on the X chromosome. There have been three reported cases of boys with Klinefelter syndrome who had similar symptoms. For male children with the normal XY genotype, this disease is fatal.
All cases of this disease are the result of sporadic mutations. That is, before this, no cases of Aicardi syndrome were identified in the proband’s family. The transmission of the defective gene from mother to daughter is unlikely. The risk that a second child will be born with this pathology is very small and amounts to less than one percent.
If we apply Mendel's laws, then the following options are possible for spouses who already have one offspring with this disease:
- stillborn boy;
- healthy girl (33%);
- healthy boy (33%);
- sick girl (33%).
The causes of the disease are currently unknown, research is ongoing.
Symptoms and signs
Aicardi syndrome usually begins with involuntary muscle spasms between the ages of four months and four years.
Other symptoms may include epilepsy, mental retardation, profound muscle weakness (hypotonia), abnormally small head (microcephaly), abnormally small eyes (microphthalmia), incomplete development of the retina and nerve at the back of the eye (coloboma), and/or rib and/or abnormalities. spine.
Children of all ages with Aicardi syndrome experience significant delays in motor development. Aicardi syndrome can be life-threatening in childhood due to complications of upper respiratory tract infections.
Most girls with Aicardi syndrome have a severe delay in psychomotor development. In the neurological status, it is often noted that there is a significant decrease in the size of the skull (microcephaly), muscle hypotonia, possibly unilateral muscle hypertension and spasticity, brisk deep tendon reflexes or impaired performance of the limbs (hemi- or tetraparesis).
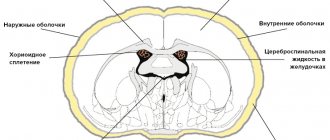
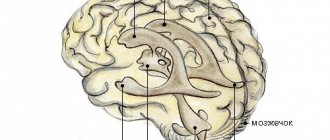
Agenesis of the corpus callosum in Aicardi syndrome is usually total, often accompanied by heterotopia of the cerebral cortex, cortical atrophy, structural asymmetry of the cerebral hemispheres, normal pressure hydrocephalus, polymicrogyria or pachygyria, choroidal cysts and papillomas, ventriculomegaly, intracerebral cysts, Dandy-Walker syndrome.
Diagnostic measures
There is no special technology for diagnosing aicardi syndrome. Carrying out diagnostics requires going through several stages, which include:
- Neurological examination . The doctor evaluates the performance of the nervous system and identifies disorders.
- Ophthalmoscopy . This research method allows you to study the fundus and retina.
- Magnetic resonance imaging. The diagnostic procedure requires the use of a constant magnetic field and radiofrequency energy. Thanks to the use of specially designed equipment, it is possible to obtain a clear image of the internal organ.
During the period of manipulation, it is possible to determine pathology in the corpus callosum, intracerebral cyst, and hemispheric asymmetry.
- Electroencephalograms . In case of aicardi syndrome, a study of the functioning of the brain is carried out, recording electrical impulses that are emitted by certain parts of the brain. The method is used in myoclinic encephalopathy to determine the type and severity of attacks.
- Computed tomography. Provides the ability to identify areas of the brain that are damaged by aicardi syndrome.
If there is a suspicion of a myoclinic type of disease, then gene studies and skeletal x-rays are recommended.
Diagnosis of aicardi syndrome requires the use of several methods, which will accurately determine the extent of organ damage.
Affected Populations
There are approximately 500 known cases of Aicardi syndrome worldwide, with a particularly large number in Japan. The syndrome occurs in children of different racial backgrounds.
According to recent studies in Sweden, the prevalence of Aicardi syndrome ranges from 2 to 15 cases per 100,000 girls. Unfortunately, similar studies have not been conducted in Russia. However, given the phenotypic diversity and diagnostic difficulties, many cases of the disease remain undiagnosed. This allows us to revise data on the true prevalence of Aicardi syndrome upward; perhaps Aicardi syndrome is a more common cause of mental retardation and infantile spasms in girls than is currently believed.
Today, it is believed that the incidence of Aicardi syndrome among all children with infantile spasms is only about 2-4%.
Causes and symptoms of the disease
The causes of myoclonic encephalopathy have not been established to date. The disease develops in newborn babies. When a child is 2-5 months old, no signs are observed. In appearance, patients do not differ from healthy children.
After reaching 6 months of age, aicardi syndrome is accompanied by convulsions and infantile spasms. The child is diagnosed with a decrease in vigorous activity and the development of stupor. The baby concentrates his gaze on one point.
Myoclonic encephalopathy is accompanied by a sharp arching of the baby’s body and straightening of the legs. Seizures are accompanied by irritability and constant crying of the baby. When seizures occur in young patients, the development of epilepsy is diagnosed.
Aicardi syndrome is accompanied by additional symptoms. The disease affects the retina of the eye, which leads to the appearance of yellowish spots. The child's visual organs are abnormally small .
The pathology is accompanied by congenital colombia, which is characterized by the presence of a gap, notch or slit in the iris. Difficulties arise during the period of feeding the baby. Myoclonic encephalopathy is accompanied by diarrhea.
Food or gastric juice enters the esophagus, which leads to the development of gastroesophageal reflux. This symptom goes away on its own and does not require medical intervention. In aicardi syndrome, muscle apathy is diagnosed.
If you monitor the patient, this will help determine the development of additional symptoms. The baby's eyebrows are located laterally . The child's nose is upturned and the incisors are excessively protruding. The angle of the nasal septum in aicardi syndrome is insignificant.
In patients, myoclonic encephalopathy is accompanied by growths and indurations on the skin - diverticula, nevi, tumor processes that develop against the background of blood vessel pathology - hemangiomas. In rare cases, abnormalities of the upper extremities are diagnosed with aicardi syndrome.
In children, the disease is diagnosed by facial asymmetry. There are clefts in the area of the upper lip and palate. The head and arms are smaller than those of a healthy child. There is a small space between the nose and lips.
Against the background of abnormalities in the eyes, patients with aicardi syndrome develop microphthalmos and rhinitis pigmentosa. Optic nerve atrophy and cataracts may develop. Patients' vision is impaired, which causes complete blindness.
Diagnostics
To date, there is no specific laboratory diagnostic test or study that would allow a diagnosis of Aicardi syndrome. To do this you need:
- neurological examination;
- ophthalmoscopy;
- electroencephalography (EEG);
- magnetic resonance imaging with and/or without contrast;
- X-ray of the skeleton.
Magnetic resonance imaging can detect agenesis of the corpus callosum, asymmetry of the cortical hemispheres, heterotopia of the cortex, intracerebral cysts, choroid plexus papilloma, etc. Cortical axons, which normally should intersect, are not formed during its agenesis and, accordingly, are not identified during neuroimaging.
Agenesis of the corpus callosum allows the lateral ventricles to extend upward into the frontal and parietal white matter. This condition is called superior translocation of the lateral ventricles into the frontoparietal regions of the brain. The third ventricle undergoes a similar upward displacement, which is one of the neuroradiological markers of agenesis of the corpus callosum. The enlarged third ventricle, moving forward and upward, pushes the anterior horns of the lateral ventricles apart; with concomitant hydrocephalus, the volume of the ventricles increases, the posterior horns expand and bend towards the midline (the “grip” shape). It is likely that the lack of supportive function of the corpus callosum is the basis for the typical feature of agenesis of the corpus callosum - enlargement of the hemispheres, third ventricle and foramen of Monroy.
Typical changes in electroencephalography in the form of hypsarrhythmia, characteristic of infantile spasms, do not occur in all patients. The most characteristic changes in the seizure EEG consist of bursts of irregular fast and slow waves lasting from 3 to 6 seconds, which are interspersed with some flattening of the basic rhythm for 5-20 seconds, and the changes are not synchronized across the hemispheres. Electroencephalography shows the “split brain” phenomenon. Since in almost half (42%) of patients infantile spasms are combined with other types of epileptic seizures, EEG data may be contradictory.
Ophthalmoscopy reveals white or yellow-white, well-demarcated, round depigmented areas.
Treatment of Aicardi syndrome
Treatment for Aicardi syndrome has not yet been developed. Symptomatic treatment is mainly used. The main treatment strategy is the relief of infantile spasms, which are often resistant to antiepileptic drugs; their treatment is complex and its effectiveness is low. Various medications are used in the highest possible doses.
Initial therapy begins with vigabatrin (Sabril) - 50-100 mg/kg/day and valproate (Depakine syrup) - 50-100 mg/kg/day. For frequent attacks, combinations of antiepileptic drugs with benzodiazepines (clonazepam) -0.25-2 mg/day, or phenobarbital (5-15 mg/kg/day) are prescribed, and suxilep 15-30 mg/kg/day can also be administered.
An alternative method is the use of corticosteroid hormones (ACTH, synacthen-depot IM, dexamethase, prednisolone) and octagam. The average dosage of prednisolone is 1-2.5 mg/kg/day, followed by a transition to the minimum maintenance dose. Hormones are prescribed in combination with basic antiepileptic drugs.
Vagus nerve stimulation can be used as a palliative surgical treatment. Since musculoskeletal defects can lead to scoliosis, physiotherapy and exercise therapy are used to prevent it, and surgical correction is possible.
Symptoms
In early-onset Aicardi-Goutiers syndrome, symptoms begin approximately 4 months after birth. The pregnancy is normal and the baby does not show any symptoms at birth. The initial signs at this stage are presented below:
- Difficulty feeding;
- Severe irritability;
- Frequent fevers for no reason;
- Erroneous sleep patterns;
- Seizures or epilepsy.
There are defects in brain development, which leads to slower head growth, deterioration of fine motor skills, and generally slower neurological development.
These symptoms are described below:
- Fear reaction even to mild stimuli;
- Only partial head control;
- Abnormalities of eye movements and infant reflexes (eg, sucking);
- Language communication also deteriorates.
Forecast
The prognosis for girls with Aicardi syndrome varies depending on the severity of their symptoms.
The prognosis for Aicardi syndrome is usually serious due to severe mental retardation and the resistant nature of seizures. Some children (up to 25%) die in the first years of life. Of the surviving children, only 25% walk independently and only 50% have self-care skills.
Life expectancy is highly variable, depending on the severity of symptoms. The average life expectancy, according to various sources, ranges from 8.3 to 18.5 years. But there is information about a 32-year-old woman with Aicardi syndrome, as well as a 49-year-old woman with a moderate form of the syndrome
Prognosis and mortality
The prognosis for Aicardi syndrome is quite unfavorable and depends on the severity of spasms and accompanying diseases:
- childhood mortality – 25%;
- survive and can walk independently – 25%;
- can take care of themselves – 50%.
Patients live from approximately 9 to 19 years. However, there are cases where affected women lived up to 32 and 49 years (moderate form of the syndrome).
Despite the rarity of the disease, a separate website is dedicated to it (www.aicardisyndrome.org). It's a great place to connect with other people living with the disease and their families. Ask questions, share experiences, and more. Here you can get information about charity events organized by the Ecardi Foundation.
Unfortunately, due to little knowledge of this disease, preventive measures against Aicardi syndrome do not yet exist.