Причины
Синдром Айкарди, вероятно, возникает в результате новой мутации в гене, расположенном на Х-хромосоме. Точный ген или генетический механизм, вызывающий синдром Айкарди, пока неизвестен. Недавно обнаруженный отчет, описывающий изменения в генах TEAD1 и OCEL1 у двух девочек с синдромом, не был подтвержден в большой группе других девочек с синдромом Айкарди. Таким образом, эти гены, по-видимому, не являются причиной синдрома Айкарди. Предполагается, что это состояние является смертельным для новорожденных детей мужского пола.
Родители женщины с синдромом Айкарди обычно не страдают. О передаче синдрома Айкарди от пострадавшей матери ее ребенку не сообщалось. Другие члены семьи также обычно не подвергаются повышенному риску.
Генетика
Айкарди синдром – это сцепленное с полом заболевание, дефектный ген располагается на Х-хромосоме. Зарегистрировано три случая, когда мальчики с синдромом Кляйнфельтера имели схожие симптомы. Для детей мужского пола с нормальным генотипом XY это заболевание является летальным.
Все случаи появления данной болезни являются результатом спорадических мутаций. То есть до этого в семье пробанда не было установлено случаев синдрома Айкарди. Передача дефектного гена от матери к дочери маловероятна. Риск, что второй ребенок родится с этой патологией, очень мал и составляет меньше одного процента.
Если применить законы Менделя, то у супругов, уже имеющих одного отпрыска с данной болезнью, возможны следующие варианты:
- мертворожденный мальчик;
- здоровая девочка (33%);
- здоровый мальчик (33%);
- больная девочка (33%).
Причины заболевания на данный момент неизвестны, исследования продолжаются.
Симптомы и признаки
Синдром Айкарди обычно начинается с непроизвольных мышечных спазмов в возрасте от четырех месяцев до четырех лет.
Другие симптомы могут включать эпилепсию, умственную отсталость, глубокую мышечную слабость (гипотонию), аномально маленькую голову (микроцефалию), аномально маленькие глаза (микрофтальмию), неполное развитие сетчатки и нерва в задней части глаза (колобомы) и/или аномалии ребер и/или позвоночника.
У детей всех возрастов с синдромом Айкарди наблюдается значительная задержка моторного развития. Синдром Айкарди может быть опасным для жизни в детстве из-за осложнений инфекций верхних дыхательных путей.
Большая часть девочек с синдромом Айкарди имеет резкую задержку психомоторного развития. В неврологическом статусе часто отмечается — значительное уменьшение размеров черепа (микроцефалия), мышечная гипотония, возможна односторонняя мышечная гипертония и спастичность, оживленные глубокие сухожильные рефлексы или нарушение работоспособности конечностей (геми- или тетрапарез).
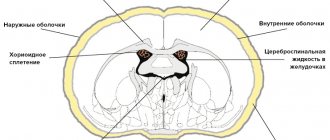
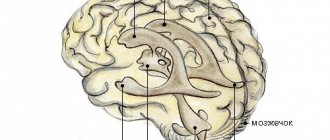
Агенезия мозолистого тела при синдроме Айкарди обычно тотальная, часто сопровождается с гетеротопией коркового вещества мозга, атрофией коры, структурной асимметрией полушарий мозга, нормотензивной гидроцефалией, полимикрогирией или пахигирией, хориоидальными кистами и папилломами, вентрикуломегалией, внутримозговыми кистами, синдромом Денди-Уокера.
Диагностические мероприятия
Специальная технология диагностики синдрома айкарди отсутствует. Проведение диагностики требует прохождения нескольких этапов, которые заключаются в:
- Неврологическом осмотре. Врачом оценивается работоспособность нервной системы и определение расстройств.
- Офтальмоскопии. Этот метод исследования позволяет изучить глазное дно и сетчатку.
- Магниторезонанской томографии. Диагностическая процедура требует применения постоянного магнитного поля и радиочастотной энергии. Благодаря использованию специально предназначенной аппаратуры обеспечивается возможность получения четкого изображения внутреннего органа.
В период проведения манипуляции обеспечивается возможность определения патологии в мозолистом теле, внутримозговой кисты, ассиметрии полушарий.
- Электроэнцефалограммы. При синдроме айкарди проводится исследование работы головного мозга, регистрации электроимпульсов, которые издаются определенными частями головного мозга. Метод используется при миоклинической энцефалопатии для определения типа и тяжести приступов.
- Компьютерной томографии. Предоставляет возможность определения участков мозга, которые повреждены синдромом айкарди.
Если имеются подозрения на миоклинический тип заболевания, то рекомендовано проведение генных исследований и рентгена скелета.
Диагностика синдрома айкарди требует использования нескольких методов, что позволит точно определить степень поражения органа.
Затронутые группы населения
Известно приблизительно о 500 случаях синдрома Айкарди во всем мире, особенно большое количество в Японии. Синдром встречается у детей с различной расовой принадлежностью.
По недавно проведенным в Швеции исследованиям распространенность синдрома Айкарди составляет от 2 до 15 случаев на 100000 девочек. В России аналогичные исследования к сожалению не проводились. Однако, учитывая фенотипическое разнообразие и диагностические трудности, многие случаи заболевания остаются недиагностированными. Это позволяет пересмотреть данные об истинной распространенности синдрома Айкарди в сторону увеличения, возможно, синдром Айкарди является более частой причиной задержки умственного развития и инфантильных спазмов у девочек, чем считается в настоящее время.
На сегодняшний день считается, что заболеваемость синдромом Айкарди среди всех детей с инфантильными спазмами составляет всего около 2-4%.
Причины и симптомы болезни
Причины миоклонической энцефалопатии на сегодняшний день не установлены. Заболевание развивается у новорожденных малышей. В возрасте ребенка 2-5 месяцев признаки не наблюдаются. По внешнему виду пациенты не отличаются от здоровых детей.
После достижения 6-месячного возраста синдром айкарди сопровождается судорогами и инфантильными спазмами. У ребенка диагностируется снижение активной деятельности и развитие ступора. Малыш концентрирует свой взгляд на одной точке.
Миоклоническая энцефалопатия сопровождается резким выгибанием тела малыша и выпрямлением ножек. Припадки сопровождаются раздражительностью и постоянным плачем малыша. При судорогах у маленьких пациентов диагностируют развитие эпилепсии.
Синдром айкарди сопровождается дополнительной симптоматикой. Заболеванием поражается сетчатка глаза, что приводит к возникновению пятен желтоватой окраски. Органы зрения ребенка имеют ненормально малый размер.
Патология сопровождается врожденной коломбой, которая характеризуется наличием щели, выемки или прорезью в радужной оболочке. В период кормления малыша появляются сложности. Миоклоническая энцефалопатия сопровождается диареей.
В пищевод попадает пища или желудочный сок, что приводит к развитию гастроэзофагеального рефлюкса. Такой симптом проходит самостоятельно и не требует медицинского вмешательства. При синдроме айкарди диагностируют мышечную апатичность.
Если наблюдать за пациентом, то это позволит определить развитие дополнительной симптоматики. Брови у малыша расположены латерально. Нос ребенка является вздернутым, а резцы – чрезмерно выступающими. Угол носовой перегородки при синдроме айкарди незначителен.
У пациентов миоклоническая энцефалопатия сопровождается наростами и уплотнениями на кожных покровах – дивертикулами, невусами, опухолевыми процессами, которые развиваются на фоне патологии кровеносных сосудов – гемангиом. В редких случаях при синдроме айкарди диагностируют аномалии верхних конечностей.
У детей заболевание диагностируется по ассиметрии лица. В области верней губы и неба имеются расщелины. Голова и руки имеют меньшие размеры, чем у здорового ребенка. Между носом и губами имеется небольшое пространство.
На фоне аномалий в глазах у пациентов развивается при синдроме айкарди микрофтальм и пигментный ринит. Возможно развитие атрофии зрительного нерва и катаракты. У больных нарушается зрение, что становится причиной полной слепоты.
Диагностика
До настоящего времени не существует специального лабораторного диагностического теста или исследования, которое бы позволило поставить диагноз синдрома Айкарди. Для этого необходимо:
- неврологический осмотр;
- офтальмоскопия;
- электроэнцефалография (ЭЭГ);
- магнитно-резонансная томография с контрастом и/или без;
- рентгенограмма скелета.
На магнитно-резонансной томографии можно обнаружить агенезию мозолистого тела, асимметрию полушарий коры, гетеротопию коркового вещества, внутримозговые кисты, папиллому сосудистых сплетений и тд.. Аксоны коры, которые в норме должны перекрещиваться, при его агенезии не формируются и соответственно не идентифицируются при нейровизуализации.
Агенезия мозолистого тела позволяет боковым желудочкам распространиться вверх, во фронтальное и париетальное белое вещество. Это состояние именуется верхней транслокацией боковых желудочков в лобно-теменные регионы мозга. Аналогичное смещение вверх претерпевает и третий желудочек, что является одним из нейрорадиологических маркеров агенезии мозолистого тела. Увеличенный третий желудочек, выдвигаясь вперед и вверх, раздвигает передние рога боковых желудочков, при сопутствующей гидроцефалии объем желудочков увеличивается, задние рога расширяются и изгибаются по направлению к средней линии (форма «ухвата»). Вероятно отсутствие поддерживающей функции мозолистого тела является основой для типичной черты агенезии мозолистого тела — расширения полушарий, третьего желудочка и Монроева отверстия.
Типичные изменения на электроэнцефалографии в виде гипсаритмии, характерные для инфантильных спазмов встречаются далеко не у всех больных. Наиболее характерные изменения приступной ЭЭГ заключаются во вспышках нерегулярных быстрых и медленных волн продолжительностью от 3 до 6 секунд, которые перемежаются некоторым уплощением основного ритма в течение 5-20 секунд, причем изменения не синхронизированы по полушариям. На электроэнцефалографии — феномен «расщепленного мозга». Так как практически у половины (42%) пациентов инфантильные спазмы сочетаются с другими видами эпилептических приступов, то данные ЭЭГ могут быть противоречивы.
При офтальмоскопии обнаруживаются белого, или желто-белого цвета, хорошо отграниченные, круглые депигментированные участки.
Лечение синдрома Айкарди
Лечение синдрома Айкарди пока не разработано. В основном применяется симптоматическое лечение. Основная стратегия терапии — купирование инфантильных спазмов, которые зачастую резистентны к антиэпилептическим препаратам, лечение их сложное и эффективность его невелика. Применяют различные медикаменты в максимально высоких дозах.
Стартовая терапия начинается с вигабатрина (сабрил) — 50-100мг/кг/сут и вальпроатов (депакин-сироп) — 50-100мг/кг/сут. При частых приступах назначают комбинации антиэпилептических препаратов с бензодиазепинами (клоназепам) -0,25-2мг/сут, или фенобарбиталом(5-15мг/кг/сут), а так же может быть введен суксилеп 15-30мг/кг/сут.
Альтернативным методом является применение кортикостероидных гормонов (АКТГГ, синактен-депо в/м, дексаметазом, преднизолон) и октагам. Средняя дозировка преднизолона 1-2,5мг/кг/сут с последующим переходом на минимальную поддерживающую дозу. Гормоны назначают в сочетании с базовыми антиэпилептическими препаратами.
В качестве паллиативного хирургического лечения возможно использование стимуляции блуждающего нерва. Так как костно-мышечные дефекты могут приводить к сколиозу, для его предотвращения используется физиотерапия, лечебная физкультура, возможна хирургическая коррекция.
Симптомы
При раннем начале синдрома Айкарди-Гутьерса симптомы появляются примерно через 4 месяца после рождения. Беременность нормальная, ребенок не проявляет никаких симптомов при рождении. Начальные признаки на этом этапе представлены ниже:
- Трудность кормления;
- Сильная раздражительность;
- Частые лихорадки без причины;
- Ошибочные режимы сна;
- Приступы или эпилепсия.
Существуют дефекты развития мозга, что приводит к замедлению роста головы, ухудшению тонких моторных навыков, вообще замедлению неврологического развития.
Эти симптомы описаны ниже:
- Реакция испуга даже на мягкие раздражители;
- Только частичное управление головкой;
- Аномалии движения глаз и детских рефлексов (например, сосание);
- Языковое общение также ухудшается.
Прогноз
Прогноз для девочек с синдромом Айкарди варьируется в зависимости от тяжести их симптомов.
Прогноз при синдроме Айкарди, как правило, серьезен в связи с выраженной умственной отсталостью и резистентным характером судорог. Часть детей (до 25%) погибает в первые годы жизни. Из выживших детей только 25% самостоятельно ходят и только 50% имеют навыки самообслуживания.
Продолжительность жизни очень вариабельна, в зависимости от степени выраженности симптомов. Средняя продолжительность жизни по разным данным составляет от 8,3 до 18,5 лет. Но имеются сведения о женщине 32 лет с синдромом Айкарди, а так же 49 лет с умеренной формой синдрома
Прогноз и летальность
Прогноз при синдроме Айкарди достаточно неблагоприятный и зависит от тяжести спазмов и сопровождающих их заболеваний:
- смертность в детском возрасте – 25%;
- выживают и могут самостоятельно ходить – 25%;
- могут самостоятельно себя обслуживать – 50%.
Больные живут приблизительно от 9 до 19 лет. Однако известны случаи, когда заболевшие женщины прожили до 32 и 49 лет (умеренная форма синдрома).
Несмотря на редкость заболевания, ему посвящен отдельный сайт (www.aicardisyndrome.org). На нем можно пообщаться с другими людьми, страдающими этим заболеванием, и их семьями. Задать интересующие вопросы, поделиться опытом и другое. Здесь можно получить информацию о благотворительных мероприятиях, организатором которых является Фонд Экарди.
К сожалению, в связи с мало изученностью данного заболевания, профилактических мер против синдрома Айкарди пока не существует.