Multiple sclerosis - symptoms and treatment
With all the diversity and variability of disorders characteristic of MS, we can distinguish its main, most common, typical form—cerebrospinal.
Cerebrospinal MS usually begins at a young age. This form is characterized by:
- a combination of pyramidal and cerebellar disorders;
- frequent damage to the optic nerves;
- sometimes transient diplopia (doubling of visible objects);
- remitting course (repeated remissions).
The development of pyramidal and cerebellar disorders is caused by the frequent formation of foci of inflammation in the lateral columns of the spinal cord, brain stem and cerebellar peduncles.
The earliest symptoms are:
- loss of abdominal reflexes;
- fatigue and weakness of the legs;
- slight intentional trembling in the hands;
- nystagmus (uncontrolled rhythmic eye movement).
Ophthalmic manifestations
Often the initial isolated disorder is visual impairment, developing as so-called retrobulbar neuritis (acute inflammation of the optic nerve). In this case, blindness or scotoma (a blind spot in the field of vision) of one or another eye occurs.
Since the process is localized not in the optic nerve nipple, but in its trunk (retrobulbar), no changes are detected in the fundus for sometimes quite a long period of time.
With significant damage to the diameter of the optic nerve, the direct reaction of the pupil of the blind eye to light is lost, while the reaction of its narrowing is preserved in the case of illumination of the other eye (friendly reaction of the pupil).
Retrobulbar neuritis in MS is characterized by loss of central visual fields (central scotoma), since plaques usually develop inside the optic nerve trunk. In contrast, with retrobulbar neuritis of syphilitic etiology or with inflammation of the paranasal cavities, a concentric narrowing of the visual fields is more often observed, since the inflammatory process develops from the outer surface of the optic nerve - from its membranes.
After a certain period of time, in MS, changes in the fundus of the eye are detected - blanching of the optic nerve nipple (atrophy). Typical for MS is the predominant blanching of the temporal halves of the optic nerves. Vision loss can be complete or partial, unilateral or bilateral. After some time, improvement usually occurs, but the process may increase. Bilateral blindness is rare.
Cerebellar and pyramidal disorders
The earliest cerebellar disorders are:
- intentional trembling in the hands, detected during the finger-nose test;
- handwriting disorder;
- nystagmus of the eyeballs when abducted to the sides.
Pronounced nystagmus (sometimes not only horizontal, but also vertical or rotatory), depending on damage to the cerebellum, speech disorders (scanned speech) and trembling of the head or torso are already a sign of an advanced process.
Other cerebellar symptoms also develop - adiadochokinesis (the inability to quickly change opposite movements - flexion or rotation), unsteady gait, etc.
Sometimes, along with pronounced symptoms of pyramidal lesions, symptoms of damage to the peripheral motor neuron are also noted: loss of tendon reflexes of the limbs, muscle atrophy.
Sensory disturbances are not as pronounced as movement disorders. A peculiar paresthesia (burning sensation, goosebumps, tingling sensation) in MS is the sensation of an electric current that occurs when sharply bending the head toward the chest, as if running down the spine, radiating to the legs and sometimes to the arms. In some cases, disturbances in the joint-muscular and vibration senses in the lower extremities are observed. Surface sensitivity is much less likely to be upset.
Of the cranial nerves, in addition to the optic nerves, the abducens and oculomotor nerves are most often affected. Paralysis of the eye muscles is usually transient and is expressed only by diplopia, which can be the initial symptom of the disease.
Lesions of the trigeminal, facial and hypoglossal nerves are common. The development of sclerotic plaques in the supranuclear parts of the corticonuclear tract can cause pseudobulbar syndrome, and in the brain stem - the development of bulbar symptoms (symptoms of damage to the cranial nerves).
Pelvic organ dysfunction is a common symptom of MS. The following violations occur:
- imperative (sudden and unsuppressable) urges, increased frequency, retention of urine and stool;
- incontinence, incomplete emptying of the bladder, leading to urosepsis - are characteristic of later stages.
Reproductive system dysfunctions are common . These problems can simultaneously be functional disorders of the pelvic organs, and can also be independent symptoms.
Mental disorders are rare. Dementia and severe forms of mental disorder are characteristic only of late periods of the disease and are caused by multiple and widespread foci in the cortex and subcortical formations of the cerebral hemispheres.
Multiple sclerosis (MS) is a chronic inflammatory autoimmune disease of the central nervous system, most common among young people, which can also occur in children. Pediatric MS, also referred to in the literature as childhood-onset MS, early-onset MS, or juvenile MS, is considered to be MS that develops before the age of 16 years (sometimes before 18 years). In 3-10% of patients with MS, the disease debuted at the age of 16 years or younger, in 1% of patients, MS began at the age of under 10 years. Pediatric R.S. has specific features, the course of the disease differs from that in adult patients [1].
Early diagnosis of MS before the age of 16 years was extremely rare, while currently there is an overdiagnosis of MS, since in childhood there is a wide range of congenital and acquired diseases with similar manifestations, which can often be mistaken for MS. To establish a diagnosis of MS in each child, it is necessary to conduct a differential diagnosis, establish the type of course and stage of the disease in order to optimize treatment methods. Accurate diagnosis could be carried out with the widespread availability of biopsy of pathological lesions to establish a clinical-pathomorphological relationship, but the high risk of biopsy of central nervous system tissue does not allow the widespread use of this method. To make a diagnosis, MRI, cerebrospinal fluid (CSF) studies are used, and less commonly, a complex study of evoked potentials and bioimmunological markers, correlating them with medical history and neurological status [2].
Common features of the pathogenesis of diseases with which differential diagnosis of MS and MS itself should be made determine their similar clinical picture and MRI data, which in turn complicates the diagnosis. In one of the latest publications devoted to the diagnosis of pediatric MS, the authors [3] analyzed in detail the clinical picture, MRI data, and response to treatment of 14 patients with mitochondrial leukoencephalopathy (ML). This rare pathology has a clinical picture that can mimic that of MS.
According to the results of numerous studies, there is a connection between mitochondrial diseases and inflammation in the central nervous system. Some primary mitochondrial diseases, such as ML, have MRI evidence of inflammation. There is modern data indicating the role of mitochondrial dysfunction in the pathogenesis of acquired demyelinating diseases, including R.S. Diagnosis M.L. in the study cohort was determined based on clinical presentation, histopathology, respiratory chain enzyme analysis, and exome sequencing.
We examined 14 patients aged 2 to 7 years, the ratio of boys to girls was 1:1. Genetic data obtained included changes in NDUFA1, NDUFV1, NDUFS2, LYRM, MPV17, BOLA3, IBA57
. Clinical features that mimicked acquired demyelinating diseases included acute onset of focal neurological symptoms associated with encephalopathy (71%), a history of pre-onset febrile illnesses (50%), partial or complete response to glucocorticosteroid (GCS) therapy, and recurrent neurological symptoms (71%) and subsequent stabilization of the condition, rather than a progressive course (85%). According to MRI, confluent foci were observed in the white matter of the brain (100%), decreased diffusion (78.5%), accumulation of contrast agent in pathological foci, involvement of the spinal cord (61.5%), lactate peak according to MR spectroscopy and cysts in the white matter of the brain (92.8%).
Some diseases that accompany MS also share the same pathogenesis of autoimmune and inflammatory processes, which may explain their comorbidity. To date, the world literature has not described many cases of concomitant diseases in children with MS. One of them is a clinical case of a teenage boy [4]. The child had multifocal neurological symptoms, including optic neuritis, as well as hypopituitarism and diabetes insipidus associated with lymphocytic hypophysitis (PH). MRI findings were consistent with the diagnosis of MS according to McDonald criteria (2010). Diagnosis R.S. was also confirmed by CSF analysis and exclusion of alternative diagnoses. The association between PH and MS has been described for the first time. PH is a rare inflammatory disease of the pituitary gland and infundibulum, most often observed in autoimmune diseases with various clinical and endocrine manifestations. On the MRI picture, PH looks like the absence of a light spot of the neurohypophysis with an enlarged cystic pituitary gland, thinning of the pituitary stalk, as well as positive dynamics during therapy with glucocorticosteroids (GCS). PH and MS have a common pathogenesis of an autoimmune process, which perhaps explains their comorbidity. Endocrine disorders such as diabetes insipidus, together with the characteristic clinical manifestations of exacerbation of MS, should encourage further research into possible concomitant inflammatory diseases involving the hypothalamic-pituitary axis [4].
We present two of our own observations of pediatric MS and cerebral palsy (CP).
Observation 1
Girl E
., 14 years old, was admitted with complaints of increased fatigue, weakness in the legs, clumsiness in the arms, periodic tremors, a sensation of “pins and needles” in the area of the fifth finger on the right hand, tremors in the legs, more pronounced on the right, periodic dizziness, inability to move without assistance help, rare urination, constipation. I was bothered by daily headaches of a pulsating nature in the temporal region, occurring mainly in the afternoon, and nagging pain in the coccyx area, radiating to the lumbar region.
History of illness: disabled child due to cerebral palsy. At the age of 3 years, the first episode of loss of consciousness occurred without the development of seizures. She was diagnosed with epilepsy and took Depakine for 2 months. An electroencephalogram (EEG) was performed: no signs of epileptic activity were recorded. EEG video monitoring: minimally expressed epileptiform activity in the central-parietal regions of the brain. The diagnosis of epilepsy was excluded. In April-May 2014, periodic headaches of a pulsating nature in the temporal region and an episode of syncope reappeared. In September 2015, there was a repeated episode of loss of consciousness without the development of seizures.
According to the mother, in November 2015, acutely after sleep, there was a feeling of numbness in the lower extremities, weakness in the legs, she could not sit up in bed or stand on her own, then frequent urination and urinary incontinence, constipation, general weakness developed, and a constant headache developed. dysarthria, emotional lability in the form of irritability, tearfulness. She was urgently hospitalized in the pediatric department at her place of residence. According to MRI of the lumbar spine: signs of right-sided scoliosis, structural changes in the spinal cord. Partial atrophy of the optic nerves was revealed. In the neurological status: mild dysarthria, tetraparesis - mild upper paraparesis to moderate in the distal parts, severe lower paraparesis. A course of symptomatic therapy was carried out. Upon discharge, the neurological status showed increased muscle strength in the legs, a decrease in the severity of sensory disorders, she began to move with bilateral support at a distance of up to 5 m, pain appeared in the coccyx radiating to the lumbar region. X-ray of the sacrum - no pathology was detected.
She was sent to the Republican Children's Clinical Hospital of Ufa, where the patient underwent an MRI of the brain with intravenous contrast: signs of demyelinating brain disease were revealed. MRI of the thoracic spine with intravenous contrast revealed focal pathology of the spinal cord. MRI of the lumbar region revealed no pathology. Electroneuromyography (ENMG) of the lower extremities: conduction disturbances of the suprasegmental type with signs of axonopathy. With EEG: moderate diffuse changes in biopotentials with rare theta waves in the central-parietal areas. During hospitalization, a clinical blood test revealed: leukocytosis 18 thousand. CSF: slightly yellow, slightly cloudy, protein 0.25 g/l, Pandi++ reaction, cytosis 36 (segmented 22, lymphocytes 14). A course of GCS therapy was carried out (dexamethasone 8 mg according to the regimen), then she received prednisolone per os
. During treatment, sensory disturbances and urinary incontinence regressed, muscle strength in the limbs increased, she began to move with bilateral support for a distance of up to 50 m, and pain in the coccyx area increased. There was clumsiness in the hands, tremors and paresthesia in the fifth finger of the right hand. She was sent for hospitalization to the Tyumen Regional MS Center to clarify the diagnosis.
Life history: born in Bashkiria, the first child of twins. The child from the first pregnancy, which was threatened with termination, was born at 32-33 weeks, the second child died at 24 days. Prematurity of the 2nd degree, body weight at birth 1400 g, asphyxia of the 2nd degree, 3-4 points on the Apgar scale. After birth, she was in the intensive care unit, on artificial ventilation for 14 days. She grew and developed with a lag in static-motor development. She started walking at 3.5 years old with bilateral support. She attended a specialized kindergarten for children with disabilities. I was worried about stiffness in my legs, more so in my left. He is registered with a neurologist and an orthopedist with a diagnosis of cerebral palsy and Little's spastic diplegia of moderate severity. She regularly received courses of rehabilitation treatment in sanatoriums and rehabilitation centers in Moscow, Evpatoria, and Anapa. In 2005, a course of autoimmune serum for atopic dermatitis. History: frequent colds, regularly received courses of antibacterial therapy. In 2012, the course of acute respiratory disease was complicated by the development of reactive gonarthritis on the left. He is registered with an ophthalmologist for hyperopia; in 2011, partial atrophy of the optic nerves was revealed. In 2013, she suffered from chickenpox.
From 7 to 14 years old she attended a comprehensive school with normal academic performance. From the 3rd grade I studied at a music school. Against the background of rehabilitation measures, an improvement in gait was noted, the distance increased to 500 m with unilateral support, and she walked around the apartment without support. Since September 2015 - distance learning. From the age of 12 - menstruation, regular. On the first day of menstruation - headache, pain in the lower abdomen.
Ultrasound of the thyroid gland: small focal formation of the right lobe. She consulted with an endocrinologist and was diagnosed with iodine deficiency goiter and euthyroidism. Ultrasound of the abdominal organs: delayed emptying of the gallbladder due to spasm of the sphincter apparatus. Ultrasound of the kidneys and bladder: no pathology was detected.
He denies hepatitis and tuberculosis. Allergological history: allergic reaction to Actovegin. She was operated on for an umbilical hernia in 2001; there were no blood transfusions. Heredity is not burdened.
Somatic status: general condition is satisfactory. Increased nutrition. Pink and white stretch marks on the skin of the chest, abdomen, and buttock area. Peripheral edema in the feet and ankle joints. Constipation. Urination is rare, free, painless.
Neurological status: general cerebral symptoms in the form of periodic headaches, dizziness. There are no meningeal symptoms. Cranial nerves (CN): the sense of smell is not impaired. Palpebral fissures, D=S. There is no anisocoria. Pupillary reactions are lively and symmetrical. Failure to bring the left eyeball outward. There is no strabismus, diplopia, or nystagmus. There is no facial pain. Palpation of the trigeminal points is painless. There are no sensory disturbances on the face or tongue. Corneal reflexes are alive, D=S. There is no facial asymmetry. There is no hearing impairment. Mild dysarthria. There is no dysphonia or dysphagia. The pharyngeal and palatal reflexes are lively and symmetrical. There are no pathological oral reflexes. Tongue in the midline. Tetraparesis: mild upper paraparesis with a decrease in muscle strength on the right to 4.0 points, on the left to 4.5 points; pronounced lower paraparesis with a decrease in muscle strength proximally to 2.5 points, distally to 1.5 points. Muscle tone is changed in a pyramidal pattern in the right arm and legs. Moderately severe spasticity in the lower extremities, mainly in the abductor muscles. Equinovalgus foot deformity. Deep reflexes from the upper limbs are alive, D
>
S
; from the lower extremities - high, clonus of the patellas, feet. The abdominal reflexes on the left are lively, but on the right they are quickly depleted. Spontaneous Babinsky reflex, Poussep, Chedok, Bekhterev, Zhukovsky, Rossolimo reflexes are evoked on both sides. Performs the finger-nose test satisfactorily. In the Romberg sample it is unstable. There are no disturbances in pain sensitivity on the trunk and limbs. Slight decrease in deep sensitivity in the toes. There are no violations of praxis or gnosis. There are no aphasic disorders.
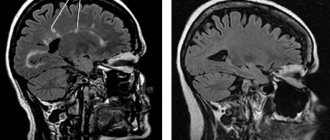
MRI of the brain with intravenous contrast: a picture of multifocal demyelinating disease of the central nervous system with signs of activity of the pathological process (presence of contrasting foci) (Fig. 1, 2).
Rice. 1. MRI of the brain. Axial and sagittal sections. T2-WI and FLAIR images show demyelination foci typical for MS.
Rice. 2. MRI of the brain with intravenous contrast. Axial slices. Foci accumulating paramagnetic contrast in the left hemisphere of the brain. In comparison with the previous MRI, positive dynamics are noted in the form of a reduction in the size of some lesions, the absence of new lesions, and a reduction in the number and size of contrasting lesions.
MRI of the cervical spine with intravenous contrast: MRI signs of multifocal demyelinating disease of the central nervous system with the presence of foci of demyelination in the cervical spinal cord without signs of activity of the pathological process in these foci (Fig. 3).
Rice. 3. MRI of the cervical spinal cord. Sagittal and axial sections. T2-weighted images show foci of demyelination typical for MS (in the spinal cord and brain stem).
Optical CT of both eyes: OU - the thickness of the nerve fiber layer is below the parameters characteristic of adults, the thickness of ganglion cells is reduced. Visual evoked potentials: conduction along the optic nerves is preserved. Computer perimetry: OD - multiple areas of severe depression and defects from 0 to 30° in all sectors of the visual fields (VF), mainly in the lower sector. OS - multiple areas of depression from 10 to 30°, mainly in the outer and lower sectors of the PZ.
Neuro-ophthalmologist: corrected vision 1.0/1.0. Fundus of the eye: the discs are pale pink, the neuro-ophthalmic rim is preserved, the expansion of the disc excavation is 0.3, the vascular bundle is slightly displaced towards the nose, the arteries tend to narrow 2:1, the course of the vessels is correct, the macular reflex is partially preserved. Conclusion: partial descending stationary atrophy of the optic nerve cannot be excluded.
EEG: bioelectrical activity of the brain is disorganized, dysrhythmic. Epileptologist: reflex syncope. There is no evidence for epilepsy. There is no need to prescribe antiepileptic drugs.
LE cells were not detected; anticardiolipin antibodies were not detected; antinuclear antibodies—screening (ANA): 0.605 (normal 0.602); antibodies to ds DNA: negative; enzyme immunoassay (ELISA) of blood for ICD: IgM, IgG negative; Epstein-Barr virus capsid protein: IgM negative, IgG positive; Blood test for HIV: negative; antibodies to aquaporin 4: less than 1:10. CSF: colorless, transparent, no cytosis, protein content is normal, culture - no microflora detected. Isoelectric focusing of oligoclonal IgG in the CSF: the pathological type of IgG synthesis was determined - oligoclonal IgG in the CSF and polyclonal IgG in the blood serum. Taking into account the medical history, MRI data of the brain, and the presence of oligoclonal IgG in the CSF, MS was diagnosed based on the MacDonald criteria (2010).
Treatment was carried out: pulse therapy with GCS (5.0 g of solumedrol per course), intravenous immunoglobulin 0.4 mg/kg, intravenous infusions of thioctacid, cytoflavin; Ceraxon, gliatilin, emoxypine, glycine, phenibut, grandaxin, omeprazole. High-dose therapy with interferon-β-1a 44 mcg (Rebif) subcutaneously 3 times a week was prescribed for a long time.
The condition at discharge is satisfactory, there is a significant positive change in the neurological status - the severity of pyramidal symptoms in the extremities has decreased, stands on toes, moves without support for a distance of up to 5 m. Muscle strength in the upper extremities proximally 5 points, distally 4.5-5.0 points ; in the lower extremities proximally 3.5 points, distally 1.5 points.
Observation 2
Girl D
., 18 years old, was admitted with complaints of visual impairment, speech impairment, inability to move independently, stand up, sit, imperative urge to urinate, difficulty urinating, constipation, periodic headaches.
From the anamnesis it is known that she was born at 32 weeks. The water-free period is during the day. At birth - height 54 cm, body weight 1600 g. Started speaking at 1.5 years. Delayed motor development - did not crawl until age 3, did not sit up, did not walk. At the age of 3 years, disability was determined with a diagnosis of cerebral palsy with tetraparesis, more pronounced in the lower extremities. CT scan of the brain: hydrocephalus. She received treatment courses at a rehabilitation center every year. As a result of symptomatic therapy, from the age of 3 she began to hold objects with her hands, crawl, and walk with bilateral support. At the age of 5 she moved independently using a walker.
In 1999, surgery was performed on both lower extremities to correct spasticity. At the age of 10, she took care of herself independently, moved within the apartment with one-sided support, and performed therapeutic exercises daily. From 6 years old - home schooling according to the general program. I finished 11th grade and took distance learning at the university.
In the fall of 2015, pain appeared in the lumbar spine, she began to spend more time in bed, increased fatigue, and began to move around with outside support. At the end of 2016, she stopped moving independently. In 2021, I couldn’t cope with the university study program and stopped studying. Since 2017, there has been a gradual deterioration in vision, memory, the appearance of an imperative urge to urinate, difficulty urinating, and episodes of urinary incontinence. In March 2021, trembling of the head and limbs appeared, spasticity in the legs increased in the morning, and she began to move around in a wheelchair within the apartment. At the end of July 2021, she noted a sharp deterioration in her condition in the form of increasing weakness in her arms, difficulty in self-care (eating), speech and swallowing problems. MRI of the brain: signs of multifocal demyelinating brain disease. An examination at the R.S. Center is recommended.
Somatic status without features.
Neurological status: involuntary laughter, euphoria, emotional lability. There are no cerebral or meningeal symptoms. CMN: sense of smell is not impaired; palpebral fissures are symmetrical; no anisocoria; pupillary reactions are lively, symmetrical; convergence is weakened on both sides with a predominance on the right; divergent strabismus on both sides; large-scale spontaneous horizontal nystagmus in the extreme leads. There is no facial pain. Palpation of the trigeminal points is painless. There are no sensory disturbances on the face or tongue. Corneal reflexes are lively and symmetrical. There is no facial asymmetry. There is no hearing impairment. Moderate dysarthria, dysphonia, mild dysphagia. The pharyngeal and palatal reflexes are reduced. There are no pathological oral reflexes. Tongue in the midline. Spastic tetraparesis: mild upper paraparesis with a predominance in the distal parts, lower paraplegia. Muscle tone is increased in a pyramidal pattern in the limbs. Pronounced spastic tone in the lower extremities. The position of the legs is forced with slight bending at the knee and hip joints. Deep reflexes from the upper extremities are lively and symmetrical; from the lower extremities, the knees are high, the Achilles are torpid, clonus of the patellas is predominant on the right. Abdominal reflexes are not evoked. Pathological foot reflexes on both sides - positive reflexes of Babinsky, Poussep, Chedok, Bekhterev, Zhukovsky, Rossolimo. The finger-nose test is performed with ataxia. Tremor of the head and torso - in a sitting position. Trunk ataxia - in a sitting position. Sits with outside support. Do not stand in the Romberg pose. There are no disturbances of superficial sensitivity on the trunk and limbs. There is a moderately pronounced decrease in deep sensitivity in the legs and feet. There are no violations of praxis or gnosis. There are no aphasic disorders. Sits in a wheelchair with support, but cannot move independently in a wheelchair. Urinary urgency, difficulty urinating, constipation.
Ophthalmologist: visual acuity - counting fingers from 1 m (in the ward). OU - large-scale nystagmus with a vertical component, pupils are symmetrical, reaction to light and convergence are weakened, limited outward movements, more on the left. Fundus of the eye: the discs are pale pink with a grayish tint, a narrow myopic cone, the vessels are narrowed, the macular reflex is preserved, the course of the vessels is correct. Conclusion: divergent paretic strabismus of the left eye, moderate myopia in both eyes, partial atrophy of the optic nerve in both eyes cannot be ruled out.
Ultrasound: bladder - volume 100 ml. The walls are thin and smooth. The content is homogeneous. No stones or additional formations were found. The orifices and lower thirds of the ureters are not dilated. The residual volume of urine is 100 ml (at the time of examination there is no urge to urinate).
Anticardiolipin antibodies were not detected, LE cells were not detected, antinuclear antibodies: within normal limits, antibodies to ds DNA: not detected, blood ELISA for ICD: IgM, IgG negative, Epstein-Barr virus capsid protein: IgM negative, IgG positive. Blood test for HIV: negative result. CSF analysis: colorless, transparent, no cytosis, normal protein; culture - no microflora detected. Diagnosis of R.S. (isoelectric focusing of oligoclonal IgG in the CSF): the pathological type of IgG synthesis was determined - oligoclonal IgG in the CSF and polyclonal IgG in the blood serum.
Treatment was carried out: pulse therapy with GCS (5.0 g of solumedrol per course), intravenous immunoglobulin 0.4 mg/kg, intravenous infusions of thioctacid, cytoflavin; Ceraxon, gliatilin, emoxypine, glycine, phenibut, grandaxin, omeprazole. High-dose therapy with interferon-β-1a 44 mcg (Rebif) subcutaneously 3 times a week was prescribed for a long time. At discharge, the neurological status showed no significant changes.
In clinical practice, a neurologist needs to take into account the possibility of the development of MS in children with other diseases of the central nervous system, including cerebral palsy, and be able to recognize new neurological symptoms against the background of the clinical picture of the existing disease.
The authors declare no conflict of interest.
Multiple sclerosis in children
Multiple sclerosis is a chronic autoimmune inflammatory disease of the central nervous system, most often diagnosed in adults, but can also affect children. Pediatric multiple sclerosis is defined as onset before age 16 years (sometimes before age 18 years). 3 to 10% of patients have an age of onset before 16 years and
Clinical signs. Children may have a wide variety of presentations, including optic neuritis, sensory, brainstem-cerebellar, and motor symptoms. The clinical course is characterized by a more aggressive and polyfocal onset, with a high rate of relapses in the early stages. In general, children tend to have a more favorable outcome after the first clinical event: slower progression (taking 10 years longer to reach the secondary progressive phase compared with adults), slower development of permanent physical disability, and better recovery from relapses (due to better plasticity). The time from onset to confirmed disability can be relatively long, but the stage of disability is reached at an earlier age [2].
Axonal damage occurs in the early stages of multiple sclerosis and leads to clinical disability. In children with inflammatory demyelinating lesions, acute axonal damage is evident. In addition to the clinical features that are typical of demyelination, multiple sclerosis is associated with significant cognitive impairment in childhood [3].
Diagnostic criteria. The International Pediatric Study Group criteria classify various acquired demyelinating syndromes that may be the first clinical sign of multiple sclerosis in children.
Clinically isolated syndrome (all required):
− Monofocal or polyfocal clinical CNS event with a suspected inflammatory demyelinating cause
− No history of demyelinating disease of the central nervous system
− No encephalopathy that cannot be explained by fever
− Inconsistency of MRI results with the diagnosis of multiple sclerosis.
Acute disseminated encephalomyelitis (all required):
− First multifocal clinical CNS event with a suspected inflammatory demyelinating cause
− Encephalopathy that cannot be explained by fever
− Three or more months after the onset of the disease, no new clinical and MRI manifestations arise
− Lesions on MRI of the brain during the acute (three-month) phase
− MRI of the brain: the lesions are diffuse, poorly defined, large (> 1–2 cm), predominantly in the white matter of the brain; rare T1 hypointense lesions in the white matter of the brain; lesions may be present in the thalamus, basal ganglia.
Pediatric multiple sclerosis (requires one of the following):
− Two non-encephalopathic clinical CNS events with a suspected inflammatory cause, separated by more than 30 days, involving more than one site
− One non-encephalopathic episode (typical of multiple sclerosis) associated with MRI findings meeting McDonald criteria; follow-up MRI shows one new lesion meeting criteria for temporal spread of multiple sclerosis
− One attack of acute disseminated encephalomyelitis, which is accompanied by a non-encephalopathic clinical episode from three months after the first symptoms and associated with new lesions on MRI
− Single acute event that does not meet criteria for ADEM, MRI findings are consistent with McDonald criteria (applies to children ≥ 12 years only).
Neuromyelitis optica (all required):
− Optic neuritis
− Acute myelitis
− Two of three auxiliary criteria: 1) on MRI of the spinal cord, a continuous lesion extending over three vertebral segments; 2) MRI of the brain does not meet diagnostic criteria for multiple sclerosis; 3) seropositive status of antibodies to aquaporin-4.
First line therapy. Currently, first-line treatment consists of interferon beta (IFNB) or glatiramer acetate (GA). The safety profile of IFNB/GA in children remains favorable. The most common side effects were flu-like syndrome (35%), liver dysfunction (26%) and injection site reaction (21%). Experience with glatiramer acetate is very limited. It has approximately the same effectiveness as IFNB [4].
Second line therapy. In children with relapses, the following drugs may be considered during first-line therapy: natalizumab, fingolimod, mitoxantrone, cyclophosphamide, rituximab, and daclizumab. However, data on the safety, efficacy and tolerability of most of these drugs are poor and are only available from small retrospective case series [5]. It is important to limit axonal damage secondary to the extensive inflammatory changes observed earlier in the disease process by initiating early treatment and delaying disability.
Literature:
- Gorman MP, Healy BC, Polgar-Turcsanyi M, et al. Increased relapse rate in pediatric-onset compared with adult-onset multiple sclerosis. Arch Neurol. 2009;66(1):54–9.
- Yeh EA, Chitnis T, Krupp L, et al. Pediatric multiple sclerosis. Nat Rev Neurol. 2009;5(11):621–31.
- Pfeifenbring S, Bunyan RF, Metz I, et al. Extensive acute axonal damage in pediatric multiple sclerosis lesions. Ann Neurol. 2015;77(4):655–67.
- Narula S, Hopkins SE, Banwell B. Treatment of pediatric multiple sclerosis. 39. Curr Treat Options Neurol. 2015;17(3):336.
- Yeh EA, Weinstock-Guttman B. Natalizumab in pediatric multiple sclerosis patients. Ther Adv Neurol Disord. 2010;3(5):293–9.