What is tuberous sclerosis
Tuberous sclerosis is a phakomatous disease of a genetic nature in which multiple organ failure is observed.
- The second most common neurocutaneous disease.
- Frequency: 1:10 LLC - 1:50 LLC per year
- Disturbance of secondary neurulation (defect in neuronal proliferation, differentiation and histogenesis) during 2-4 months of pregnancy
- Autosomal dominant type of inheritance
- Currently, two gene loci have been described: 9^34 and 16p13.
Description
Tuberous sclerosis is a hereditary disease manifested by disturbances in the stable functioning of the central nervous system, which cause attacks of epilepsy and are characterized by a low level of intellectual abilities. The disease provokes the development of rashes on the surface layer of the skin of a polymorphic nature, the formation of malignant neoplasms and non-tumor processes in the somatic parts. As diagnostic measures, medical staff at clinics resort to the use of magnetic resonance imaging, computer studies, and EEG to identify disorders of the nervous system.
Additionally, ophthalmological testing is carried out, assessing the condition of internal objects of the body using ultrasound equipment, radiography and sigmoidoscopy. Treatment involves the use of antiepileptic therapy, neuropsychological adjustments, monitoring and timely surgical removal of tumors.
Quick Facts
Tuberous sclerosis (TS) is a serious disease of hereditary origin, which manifests itself with dangerous symptoms such as deformation of the surface of the epidermis, convulsions, mental retardation and the formation of tumors. The anomaly is classified as phacomatous. It is a fairly rare disorder. According to average statistical data, TS is diagnosed in a ratio of 1:30,000 of the population, and among infants the rate drops to 1:8,000 cases. Deviation from the norm is familial and isolated (up to 70%) in nature.
History of discovery
Friedrich Daniel von Recklinghausen, a German scientist (pathologist) in the mid-19th century, became the first person to give a complete description of the disease. Later, the French scientist Bourneville described in detail the morphological changes that occur in the patient’s brain with such a lesion. He gave the anomaly a name. And in 1890, the dermatological doctor Pringle analyzed the symptoms of faces using angiofibre. As a result, deviation goes by several names. In the medical literature you can find: Tuberculous sclerosis, Bourneville-Pringle disease.
Main sources of appearance
The disorder is distinguished by the hereditary nature of the lesion. Most cases are caused by the development of mutation reactions, and 30% are caused by autosomal dominant transmission of genetic aberrations that are present in the mother and father. However, scientists are still conducting research to study aspects of biochemical factors. A morphological anomaly is the growth of particles of the glial plane of the cerebral zone. In structure, they are represented by large cells with an abnormally enlarged center and many processes. Subependymal nodes usually provoke the progression of giant cell astrocytoma. According to information provided to the researchers, cerebellar damage is observed in 10% of situations. Disturbances are observed in the area of the optic nerve and peripheral areas of the retina.
Clinical manifestations
Typical symptoms:
- "White spots" of melanin hypopigmentation
- Facial angiofibromas: reddish nodules, initially the size of a grain of rice, later larger butterfly-shaped lesions on the cheeks, nasolabial folds and nose
- “Shagreen” plaques: skin lesions of various sizes with a “skiny” proliferation of connective tissue, which are localized mainly in the lumbosacral region
- Generalized convulsive seizures.
- Tuberous sclerosis in children has its symptoms from the first years of life - mental retardation, convulsive attacks in the form of myocloic seizures. The peak occurs during puberty. Then the disease acquires the symptoms indicated above.
Treatment of tuberous sclerosis in Germany
Since tuberous sclerosis is a genetic pathology, today there is no etiological and pathogenetic treatment for this disease. However, properly selected symptomatic therapy can significantly improve the patient’s quality of life. German specialists use medical and surgical methods to treat this disease. As a rule, in most clinical cases it is necessary to combine therapy.
Conservative treatment
- Anticonvulsants . Very often, Bourneville disease manifests itself as tumors in the brain that cause seizures. In such cases, the attending physician, together with a neurologist, selects an anticonvulsant for the patient.
- One of the main manifestations of the disease is mental retardation. Therefore, from early childhood, when the diagnosis is already known, the patient is prescribed neurostimulating and neurotropic drugs to prevent the progression of the pathological process.
- When intracranial pressure increases, osmotic diuretics and dehydration therapy are prescribed.
- Conservative treatment of heart failure that occurs due to rhabdomyoma. For this purpose, beta blockers, ACE inhibitors, calcium antagonists and cardiotropic drugs are prescribed.
- For respiratory cysts, progesterone preparations .
Medicines for tuberous sclerosis should be taken continuously, for life and under the strict supervision of the attending physician.
Surgery
The surgical method is used in the event of neoplasms that threaten the patient’s life, there is a high risk of their malignancy, and they also cause great discomfort to the patient.
German clinics are equipped with modern equipment that allows them to perform minimally invasive interventions.
Typically, patients with tuberous sclerosis undergo the following surgical procedures:
- Removal of brain tumors is performed when conservative therapy does not improve the patient’s condition. In German clinics it is possible to perform such operations under visualization control, which significantly reduces the risk of postoperative complications;
- Removal of polyps of the gastrointestinal tract. In cases where neoplasms in the intestines lead to persistent digestive disorders and there is a risk of malignancy, it is necessary to remove the polyps. Such surgery can be performed using modern endoscopic equipment. In this case, the patient will not feel any discomfort or pain during the procedure (which takes place under general anesthesia) and after it. Removed elements are necessarily sent for histological examination;
- If renal bleeding occurs, nephrectomy must be performed. If the patient develops kidney cysts, they are punctured;
- Skin defects that are localized on the face can be removed using dermabrasion. This is a modern method, which is based on the removal of the surface layer of the epidermis. The disadvantage of this method is the frequent recurrence of the pathological process. Therefore, German specialists at clinical institutions more often remove tumors on the face using an argon laser;
Being treated in highly specialized clinics in Germany, patients have many advantages. Because German specialists, using modern treatment methods, can provide patients with tuberous sclerosis with a full life.
Which method of diagnosing tuberous sclerosis to choose: MRI, CT, angiography
Selection method
- MRI
Is MSCT of cerebral vessels informative in Bourneville-Pringle disease?
Tuberous sclerosis shows calcified nodules or calcified subependymal hamartomas on imaging.
What will MRI images of the brain show in tuberous sclerosis?
Cortical nodes:
- Usually multiple
- Most often found in the supratentorial region
- In newborns, they are hyperintense in relation to the adjacent, still unmyelinated white matter on T1-weighted images and hypointense on T2-weighted images
- In older children, the center of the nodes appears hypointense relative to the white matter on T1-weighted images and hyperintense on T2-weighted images.
- In the mature brain, lesions may be isointense on T1-weighted images, but almost always hyperintense on T2-weighted images
- Calcifications may be present
- FLAIR and magnetization transfer techniques provide more diagnostic information regarding the size and number of lesions.
Subependymal hamartomas:
- Subependymal nodes are most often found along the ventricular contour of the caudate nucleus, just posterior to the interventricular foramen of Monro, but can also be located in other areas
- In newborns, the lesions are hyperintense in relation to the adjacent, still unmyelinated white matter in T1-weighted images and hypointense in T2-weighted images
- As the brain matures, lesions increasingly appear isointense to white matter on T1- and T2-weighted images.
- The nodes become calcified, but not before the end of the first year of life
- Contrast enhancement may be observed but is not clinically significant.
Subependymal giant cell astrocytomas:
- Usually located near the interventricular foramen of Monroe
- Typically larger than 12mm
- Often lead to occlusive hydrocephalus
- Always accumulate KS
- May become calcified.
White matter lesions:
- The signal pattern is identical to that of the cortical nodes
- FLAIR and the magnetization transfer method provide more diagnostic information.
Tuberous sclerosis (TS) is a rare genetic multisystem disease that usually appears shortly after birth. The disease can cause a wide range of signs and symptoms and is associated with the formation of benign tumors in various organ systems of the body. The skin, brain, eyes, heart, kidneys and lungs are often affected. These tumors are often called hamartomas. Hamartoma is a nodular benign tumor-like formation, which is a tissue developmental abnormality. The term was proposed in 1904 by the German pathologist Eugen Albrecht (1872-1908) when describing liver dysembryoplasia. A hamartoma consists of the same tissue components as the organ where it is located. Moreover, it is distinguished by its abnormal structure and degree of tissue differentiation. Hamartomas are not cancerous; they usually do not metastasize or spread to other parts of the body. However, these abnormal growths can grow in size and damage the affected organ system. The number, size, and specific location of these growths in people with tuberous sclerosis can vary widely, and therefore the severity of the disease can also vary widely. Tuberous sclerosis occurs as a result of mutations in a gene or genes, which may occur spontaneously (sporadic) for unknown reasons or be inherited as an autosomal dominant trait. Most cases are new mutations (de novo) with no family history of the disease.
Story
Désiré-Magloire Bourneville (1840–1909) was a French neurologist who is notable for the original description of tuberous sclerosis ("Bourneville's disease") in 1880.
John James Pringle (1855-1922) was a Scottish dermatologist who also studied this disease, leading to it being referred to as "Bourneville-Pringle disease" in some books.
Heinrich Vogt (1875-1936) was a German neurologist who is famous for establishing three pathognomonic clinical features of tuberous sclerosis, which became known as the "Vogt triad".
Etymology
The frequency of TS in the population is 1:10000 (in newborns – 1:6000). The estimated number of patients with TS in the Russian Federation is about 7,000 people, so TS is classified as a rare (orphan) disease. The disease is believed to affect about 2 million people worldwide. Men and women are affected in equal numbers, and the disease occurs in all races and ethnic groups.
Genetics
Mutations in the TSC1 or TSC2 gene can cause tuberous sclerosis complex. The TSC1 and TSC2 genes synthesize the proteins hamartin and tuberin.
- TSC1: encodes hamartin, on chromosome 9q32-34
- TSC2: encodes tuberin on chromosome 16p13.3
Inside the cell, these two proteins likely work together to help regulate cell growth and size. Proteins act as tumor suppressors, which typically prevent cells from growing and dividing too quickly or uncontrollably.
People with tuberous sclerosis complex are born with one mutated copy of the TSC1 or TSC2 gene in each cell. This mutation prevents the cell from producing functional hamartin or tuberin from the altered copy of the gene. However, the other, normal copy of the gene usually produces enough protein to effectively regulate cell growth. For some types of tumors to develop, a second mutation involving a different copy of the TSC1 or TSC2 gene must occur in certain cells during a person's lifetime.
When both copies of the TSC1 gene are mutated in a particular cell, that cell cannot produce any functional hamartin; cells with two altered copies of the TSC2 gene are unable to produce any functional tuberin. The loss of these proteins allows the cell to grow and divide uncontrollably, forming a tumor. Loss of hamartin or tuberin in different cell types leads to the growth of tumors in many different organs and tissues.
In rare cases, people with tuberous sclerosis complex do not have a mutation in the TSC1 or TSC2 gene. Or there are people with mutations in both TSC1 and TSC2. Research suggests that in these cases, the condition may be caused by a random mutation in the TSC1 or TSC2 gene that occurs very early in development. As a result, some cells in the body have a normal version of the gene, while others have a mutated version. This situation is called mosaicism.
Signs and symptoms
Damage to the central nervous system:
Typical features are the presence of tuberos in the cerebral hemispheres, subependymal nodules on the walls of the lateral ventricles, and subependymal giant cell astrocytomas (SEGA) near the foramen of Monro.
Skin manifestations:
Hypopigment spots
are one of the most common skin manifestations of tuberous sclerosis (Ash leaf spots). Also, patients often have a gray strand of hair from early childhood.
Cardiac changes:
In infancy, 50-60% of patients with TS have signs of heart disease, mainly manifested by the presence of rhabdomyoma.
Ophthalmological changes:
at least 50% of patients have ocular abnormalities; some studies report prevalence as high as 80%; these lesions are retinal astrocytomas, which tend to calcify over time.
Pulmonary changes:
studies have revealed cystic changes in lung tissue in 40% of women with TS.
Renal changes:
First of all, renal tissue is usually affected by cysts, since the complex of mutations in tuberous sclerosis is close to the complex of mutations in polycystic disease. Lipomas and angiomyolipomas are also often detected in the kidneys. Patients may also develop renal cell carcinomas.
Dental changes:
fibroids in the oral cavity occur in 70% of adults.
Gastrointestinal disorders:
hamartomas and polyposis of the stomach, intestines and colon are possible. Liver disorders: Liver cysts and liver AML, usually asymptomatic and nonprogressive, have been reported in 24% of patients with TS, with a notable female predominance (female to male ratio 5:1)
Skeletal disorders:
sclerotic and hypertrophic lesions of bone tissue can be accidentally detected during radiography.
Diagnosis of tuberous sclerosis
TS is diagnosed using genetic testing and a series of clinical and laboratory tests, which include:
- MRI of the brain
- Electrocardiogram
- Echocardiogram
- Kidney ultrasound
- Eyesight check
- EEG
- Examination of the patient using a Wood's lamp (emitting ultraviolet light)
Seizures or developmental delay are often the first sign of TS. There is a wide range of symptoms associated with this disease, and an accurate diagnosis will require a wide range of tests.
Radiological features
The method of choice for examining pinettes with tuberous sclerosis is MRI; with this type of research, the two anatomical areas that are most informative for us are the brain and the kidneys.
Brain
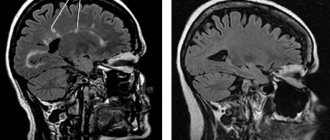
For young children, the most typical changes in the still incomplete myelination processes are linear foci on T2WI detected along the migration paths of the cerebral hemispheres. These changes can be combined with cortical and subcortical lesions (tubers), or be the only manifestation in an MRI study. The presence of cortical and subcortical tubers is typical for all age groups of patients. Cysts can form in their projection, and they can also be partially or completely calcified. Their size and number vary individually for each patient.
The most typical location is the cerebral hemispheres, but in 15% of patients the tubera may be located in the cerebellum. Tubers are glioneuronal hamartomas and consist of disorganized white matter cellular tissue.
On the walls of the lateral ventricles, subependymal nodes are detected, which often accumulate calcium. If these nodes are located near the foramen of Monroe, then these nodes are considered to be astrocytomatic. Subependymal giant cell astrocytoma (SEGA) is located near the foramen of Monroe (please note that this term is used for formations larger than 5 mm). Contrast enhancement must be used to correctly assess the size of astrocytomas. Estimating the size of the SEGA is very important for assessing the dynamics of the disease, therefore, in the study protocol it is necessary to indicate not only the three maximum transverse sizes of the formation, but also calculate the volume of the tumor. The volume is calculated using the formula for determining the volume of an ellipsoid (given by the following formula: 4/3pi*abc
). Size estimation is made only on contrasting sequences.
Urinary system (Kidneys)
MRI assessment of renal tissue is also very important for diagnosis and assessment of the course of the disease. Ultrasound and MRI reveal cysts of different sizes in the kidneys, lipomas and angiomyolipomas. Data from literature sources place the main emphasis on identifying cysts in the kidneys, due to the proximity of mutations in tuberous sclerosis and polycystic kidney disease. However, this is not entirely true; the main emphasis should be on identifying angiomyolipomas (AML). There are two main types: low-fat AML and epithelioid AML. Since for patients with tuberous sclerosis, malignant tumors (carcinomas) can also form. Carcinomas develop on their own, but they are very similar to low protein AML, so these AMLs must be described separately to avoid missing the formation. Classic angiomylipomas appear hyperintense on T1- and T2-weighted images and hypointense on T1-weighted images.
Cardiovascular system
Echocardiography – Some patients with TS have rhabdomyomas. Rhabdomyoma is a type of benign myocardial tumor that is considered the most common heart tumor. Develops most often in children under 15 years of age. Rhabdomyoma can be localized in any part of the heart, excluding the valve apparatus, most often in the ventricles, and often has an intracavitary growth pattern.