Lissencephaly, schizencephaly, porencephaly: causes, symptoms, prognosis
Smoothing of the convolutions of the cerebral cortex (agyria) is formed in utero.
The pathogenetic mechanism for the development of the nosology is a violation of embryogenesis caused by the pathology of the spread of neuroblasts from the primary neural tube. Smoothing of the surface is accompanied by a decrease in mental and intellectual activity. Concomitant neurological disorders gradually increase. The disease is fatal in infancy.
The absence of cerebral convolutions excludes the optimal functioning of the second signaling system, which exists only in humans. Folding increases the functional surface of the brain.
Even early detection using brain MRI cannot save the life of a child with type 1 or type 2 lissencephaly.
During pregnancy, congenital malformations of cerebral structures are determined by ultrasound examination (ultrasound). The reliability of the procedure is 60%, so sometimes congenital malformations of the brain are detected after MRI, although intrauterine monitoring of pathology does not determine pathological conditions.
An example of a magnetic resonance imaging report:
- Bilateral ventriculomegaly;
- Increased liquor spaces;
- Smoothness of the cerebral convolutions;
- Incomplete absence of folds in the parietal and frontotemporal regions;
- Dilatation of the lateral ventricles.
The nosology is often combined with the stigmas of cerebral dysembryogenesis - macrocephaly, agyria, microcephaly.
Medical sources claim that pathology can be detected by ultrasound after the twenty-sixth week of pregnancy.
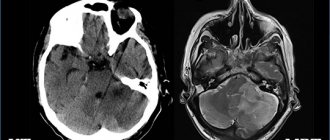
MRI: normal and lissencephaly
Lissencephaly type 2 is alternatively called Norman-Roberts syndrome. Canadian scientists who discovered the disease consider the mutation of the “RELN” gene to be the main cause of the disease.
The DNA region responsible for nosology encodes the formation of the “Reelin” protein. The syndrome is characterized by many clinical changes:
- Lymphedema (lymphedema);
- Epileptic seizures;
- Sloping forehead;
- Thickening of the cerebral cortex;
- Agyria (smoothness of the cerebral sulci);
- Macrocephaly;
- Microcephaly.
Perinatal ultrasound detects most cases of the disease.
Lissencephaly
The main common cause of all types of lissencephaly is a disruption in the process of migration of neuronal precursor cells (neuroblasts) from the anterior parts of the neural tube to the future cerebral cortex. As a result, instead of a complex folded structure, which has six layers, a smooth or much less grooved cortex is formed, consisting of 2-4 layers (depending on the form of the disease). Since the human cerebral cortex is responsible for cognitive functions, it contains a huge number of nerve centers and extensive associative zones, lissencephaly leads to severe disorders. In addition, with some types of mutations that cause this condition, abnormalities of other organs and tissues may develop, which further aggravates the patient’s condition.
The most common types of lissencephaly are caused by defects in the PAFAH1B1 gene, also known as LIS1, located on chromosome 17. Many geneticists note that for severe underdevelopment of the cerebral cortex and Miller-Dieker syndrome to occur, it is not point mutations in LIS1 that are required, but large deletions involving hundreds of nitrogen base pairs. Often, surrounding genes are also damaged, which causes various phenotypic manifestations of this type of lissencephaly. The LIS1 gene encodes an intracellular subunit of a complex enzyme that takes an active part in the migration of neuroblasts. Defects in the structure of this subunit lead to disruption of this process and the development of this disease.
Another form of lissencephaly is caused by a mutation in the DCX gene, located on the X chromosome, so the inheritance of this type of pathology is sex-linked. The protein resulting from the expression of this gene is involved in the formation of a special type of microtubules, which are produced only by neuroblasts and are necessary for them to form connections between cells. Disturbances in the structure of DCX lead to the synthesis of a defective protein, which provokes lissencephaly. Another relatively studied variant of this condition, also X-linked, is caused by mutations in the ARX gene, which is a transcription factor for other genes. It controls the development of the cerebral cortex, pancreas and genital organs, which is why ARX defects are manifested by numerous defects of these organs, including lissencephaly.
Another common variant of lissencephaly is a form of the disease caused by a mutation in the RELN gene, located on chromosome 7. Defects of this gene lead to the so-called Norman-Roberts syndrome, which, among other things, is accompanied by a pronounced violation of the folding of the cerebral cortex. The RELN gene encodes the sequence of the glycoprotein Reelin, which takes an active part in the formation of nervous tissue, and also controls the formation of new dendrites and the functioning of long-term memory in adults. It was possible to identify another gene leading to the development of lissencephaly - TUBA1A, which is localized on chromosome 12. It encodes a certain component of the neuronal cytoskeleton; its defect leads to the inferiority of neuroblasts, which disrupts the process of their migration in the embryonic period.
Causes of lissencephaly
The formation of the intracerebral cortex in the fetus goes through several stages of development. First, neurons divide. Gradually, nerve cells migrate from the original matrix to their permanent locations. The remaining stages of embryogenesis are accompanied by the specialization of cell layers responsible for anatomical structures.
The complex process of brain formation must take place under the strict control of biochemical mechanisms. Any external intervention disrupts intrauterine development.
The main causes of lissencephaly:
- Intrauterine hypoxia (oxygen starvation);
- Viral infections;
- Chromosomal abnormalities (defects in the formation of the Reelin protein on chromosome 7).
Intrafamily cases of transmission of the disease are quite rare. If the nosology occurs in several family members, genetic counseling is required.
The average prevalence of type 1 nosology is 11% per million newborns. Most situations are caused by genetic defects of the LIS1 gene. In second place is cerebellar hypoplasia due to a defect in the TUBA1A gene.
After hereditary anomalies, viral infection in the first trimester of pregnancy is in second place.
Main types of lissencephaly
There are over twenty morphological forms of the disease:
- Classic form (type I) with LIS1 mutation, Miller-Dieker syndrome;
- Form with hereditary defect DCX;
- Isolated variety without mutations;
- Agenesis of the corpus callosum due to an abnormality of the X chromosome;
- Cerebellar hypoplasia with reelin coding disorder (Norman-Roberts syndrome);
- Microlissencephaly;
- Cobblestone appearance with Fukuyama syndrome, Walker-Warburg syndrome, musculocular-brain form.
Muscular dystrophy with Fukuyama mental retardation is transmitted in an autosomal recessive manner. Hereditary myotonia has a progressive course.
Concomitant diseases and disorders
This developmental defect may be accompanied by Miller-Dieker or Walker-Warburg syndrome.
Miller-Dieker syndrome is characterized by classic lissencephaly, with this syndrome affecting the facial
Walker-Warburg syndrome
muscles and other possible defects appear that can only be found in people with this syndrome.
A feature of the syndrome is the loss of several genes on chromosome 17. The loss of the PAFAH1B1 gene is responsible for lissencephaly; this is a fact proven by scientists.
But if the YWHAE gene is lost, lissencephaly can become more complicated. The consequences of loss of other genes in this syndrome are not yet known.
Walker-Warburg syndrome is a very rare congenital muscle dystrophy associated with disruption of the brain and facial muscles.
Features of schizencephaly, porencephaly, holoprosencephaly
Bilateral cleft of the cerebral hemispheres is formed at the stage of embryogenesis. The morphological structure of the formation can be open or closed.
Unilateral localization of pathology on MRI images may resemble a cyst. Along the cleft there are pathological areas of cerebral parenchyma - microgyria, zones of increased repeated activity, spastic paralysis.
Porencephaly is characterized by the formation of cystic cavities within the cerebral parenchyma. The causes of the pathology are hemorrhage inside the brain, heart attacks, strokes. The typical location of the cyst is the Sylvian fissure.
In rare cases, porencephaly is characterized by the formation of abnormal passages between the cystic cavity and the ventricular spaces. The nosology is often characterized by other stigmas of disembryogenesis:
- Microcephaly;
- Encephalocele;
- Agiriya.
A child with a developmental defect experiences neurological disorders:
- Oligophrenia (mental retardation);
- Epilepsy attacks;
- Atrophic changes in the optic discs;
- Tetraparesis.
Concomitant changes complicate the clinical picture:
- Arterial and venous hemorrhages;
- Intracranial hypertension (in case of impaired circulation of cerebrospinal fluid);
- Cerebral infarctions;
- Hemiparesis;
- Focal epileptic foci.
Pseudoporencephalic cysts often have a unilateral (one-sided) location. They are often combined with defects of the central nervous system and defects in cell migration.
Schizencephaly on MRI
Holoprosencephaly - what is it?
The disease occurs due to the pathological division of the chemical substance prosencephalon.
Classification of nosology by severity:
- Lobarnaya;
- Semi-lobar;
- Alobar.
The heaviest is the last form. With it, a number of congenital anomalies are observed:
- Underdevelopment of the premaxillary bone;
- Sinophthalmos;
- Cebocephaly.
Against the background of multiple anomalies, parts of the face are difficult to distinguish. Associated neuronal migration defects:
- Synostosis (fusion of nerve ganglia);
- Presence of one cerebral ventricle.
The etiological mechanisms of hereditary defects have not been established. Holoprosencephaly is fatal in most cases, but the incomplete form can lead to disability.
Symptoms of lissencephaly
Clinical manifestations of the nosology are determined immediately after the birth of the child. Not only genetic defects are noted, but also swallowing disorders in the baby, increased muscle tone, and developmental delay from peers.
Symptoms of lissencephaly in a child 2-5 months old:
- Oligophrenia;
- Mental development disorder;
- Myoclonus;
- Increased blood pressure.
At first, mental retardation is not evident. After the first year of life, disorders of psycho-speech skills are observed. Children are irritable, anxious, and muscle activity is reduced.
When compared with peers, there is a significant lag in development, formation of the muscular system, innervation of internal organs, and the pelvis.
With a mild course of the disease, the child can live until adolescence. External anomalies limit social communication. Patients rarely live to be eighteen years old, but there are about twenty types of pathology. Manifestations of the first type:
- Macrogyria;
- Schizencephaly;
- Microcephaly;
- Four-layer structure of the cortex;
- Pontine hypoplasia;
- Underdevelopment of the cerebellum.
It is difficult to determine lissencephaly in the absence of positive results of intrauterine ultrasound. The clinical picture appears after birth. Muscle paralysis and cramps appear immediately or during the first year.
In most cases, ultrasound examination will help establish the diagnosis of lissencephaly. A doctor may order magnetic resonance imaging for a pregnant woman and baby. MRI is harmless, but is not performed in the first trimester due to the lack of practical information about the effect of the magnetic field on the fetus.
Treatment methods for lissencephaly in Israel
Lissencephaly is a rare complex disorder of brain function that is caused by abnormalities in fetal neurogenesis between 21 and 24 weeks of pregnancy. Most neurons cannot reach the outer part of the cerebral cortex and remain under the cortical plate. As a result of abnormal development, the newborn's brain is almost completely devoid of convolutions and has a smooth surface. Children with this pathology are usually significantly delayed in development, but everything is determined in each specific case individually.
Today, more than 20 different types of lissencephaly have been classified, which differ in clinical manifestations and characteristics of gene mutations. Treatment methods for lissencephaly in Israel are based on the relief of symptomatic manifestations, which include seizures, respiratory dysfunction, spasticity, swallowing difficulties, etc.
In a typical case, the patient receives comprehensive treatment. The team of doctors may include neurologists and pediatricians, neonatologists and geneticists, and other specialists.
Approximately 90% of children with lissencephaly suffer from epileptic seizures. To prevent them, patients receive medications from the group of anticonvulsants
. Today, many effective drugs have been developed - these are some tranquilizers and synthetic analogues of GABA, derivatives of succinic acid, etc. Anticonvulsants from the group of analogues of GABA - gamma-aminobutyric acid, a neurotransmitter involved in many processes occurring in the nervous system - are especially promising. Modern analogues of GABA can not only prevent epileptic seizures, but also stabilize the patient’s mood, relieve spasticity, reduce anxiety and fight depression.
Sometimes patients with lissencephaly are diagnosed with hydrocele. Various types of bypass surgery
- creating artificial pathways to drain excess cerebrospinal fluid from the ventricles of the brain into the abdominal or thoracic cavity.
Endoscopic procedures
are also widely used - ventriculocisternostomy, aqueductoplasty, etc.
If the patient has problems swallowing, a gastrostomy
- installation of a system that allows food to be introduced into the stomach, bypassing the esophagus. The clinic’s specialists are proficient in several techniques for performing gastrostomy, including percutaneous endoscopic, in which instead of an incision in the abdominal wall, a puncture is made and all the necessary microsurgical instruments are inserted through it.
Physiotherapy and exercise therapy may be prescribed to prevent muscular dystrophy.
.
Also, the patient, depending on the individual clinical picture, may undergo treatment aimed at relieving negative manifestations from the cardiovascular, urinary, respiratory systems, etc.
The first signs of lissencephaly in newborns
Early detection of nosology helps to prolong the baby’s life, but it cannot save him from death. The first signs in newborns:
- Small head;
- Apathy, lethargy;
- Wide interocular distance;
- Epileptic convulsive syndrome;
- Hypertonicity of muscles;
- Increased width between the lungs and kidneys;
- Activation of pathological reflexes;
- Sloping frontal part.
During the first year of life, other manifestations appear - speech disorders, urinary incontinence, increased breathing and heart rate.
Lethal pterygum (Norman-Roberts) syndrome occurs in type 1 lissencephaly. A number of manifestations are added to the above-described manifestations:
- Difficulty sitting;
- Inability for the baby to maintain a vertical position;
- The appearance of generalized muscle twitching;
- Anomalies of the craniofacial region (wide eyes, sloping forehead, bumps on the back of the head);
- Ocular nystagmus;
- Decreased muscle tone.
Cerebellar ataxia is characterized by a lack of coordination. The child cannot maintain balance or maintain an upright position. Parents first pay attention to the difficulties of planting and maintaining a horizontal position.
Diagnosis of lissencephaly
The diagnosis is made immediately after birth using ultrasound, CT and MRI. Manifestations can be detected in utero from 20 weeks. Detection of the slightest disturbances in the development of the fetal brain requires additional diagnostics. Depending on the degree of damage, a decision is made on the possibility of terminating the pregnancy. If hereditary forms are suspected, genetic counseling is carried out for all family members.
Late verification of pathology requires constant conservative treatment. Therapy for severe forms is carried out in a hospital under the supervision of qualified doctors.
The main types of diagnosis of lissencephaly:
- Magnetic resonance imaging (MRI of the head) shows soft tissue. The study reveals cerebral anomalies, cysts, fluid accumulations, inflammatory foci;
- Intrauterine ultrasound of the head is performed at 22-27 weeks, when the process of furrow formation is observed;
- Computed tomography (CT) helps to verify changes in gray and white matter, additional solid formations. The examination leads to radiation exposure of tissues, so for children it is performed according to strict indications;
- Electroencephalography (EEG) verifies foci of increased brain activity and areas of parenchymal hyperactivity.
The most reliable study is MRI for lissencephaly in newborns, which allows you to determine the smallest anomalies.
Types of development
Let's consider pathology based on 4 types of development.
Type one
The baby's brain can be compared to the brain of a fetus at 23 weeks of development. The cortex consists of four layers of neurons, pachygyria and agyria develop.
Peculiarities:
- smooth outer part of the cerebral cortex;
- the volume of white matter in the brain is greatly reduced;
- ribbon heterotopia, which is separated from the bark by a white stripe;
- trunk hypoplasia.
Type two
The cerebral cortex is much thicker than it should be, without a normal layer structure, there are disturbances in the vascular tree; hydrocephalus is present.
Peculiarities:
- smooth bark surface;
- hypoplasia;
- the boundary between white and gray matter is blurred;
- thickening of the cortex;
- agenesis of the corpus callosum;
- in some patients there is an encephalocele in the occipital region.
Type three
Peculiarities:
- white matter in smaller quantities than needed;
- hypoplasia;
- convolutions begin to stand out, but this is hardly noticeable.
Type four
Peculiarities:
- stop myelination;
- the brain is very small in size;
- the number of neurons is 35% of normal;
- Some convolutions and grooves of normal thickness are visible on the cortex.
In some cases, another classification of lissencephaly is used, it is based on the severity of the disease:
- the heaviest class, total agyria , there are no convolutions;
- there is a small number of folds on the occipital and frontal stripes, diffuse agyria ;
- combination of pachygyria and agyria;
- diffuse pachygyria;
- anterior pachygyria and heterotopia;
- heterotopia under the bark.
The first and fourth degrees are very rare, the second is found in children with Miller-Dieker syndrome. The most common is a combination in the third degree; usually this pathology consists of posterior agyria and frontal pachygyria.
Type one, that is, classic, occurs in 12 people per million births.
Video on the topic: