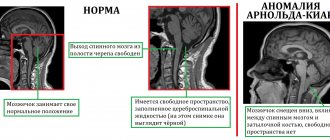
In clinical practice, there are often difficulties in making a differential diagnosis of multiple sclerosis (MS) and other diseases with damage to the central nervous system, in which similar clinical and tomographic signs are observed, especially at the onset of the disease [1, 28, 49]. Thus, S. Watanabe et al. [53] followed a woman for a long time who, at the age of 38, gradually developed slowly progressive spastic monoparesis of the left leg, and she had difficulty walking; At 40 years of age, clinical examination revealed slow speech, vertical nystagmus, spasticity in the arms and legs and monoparesis of the left leg, increased mandibular reflex, hyperreflexia of tendon reflexes, and positive bilateral Hoffmann and Babinski reflexes. Laboratory examination revealed positive titers of antinuclear antibodies and anti-SS-A/Ro antibodies. Cerebrospinal fluid (CSF) showed oligoclonal bands and elevated IgG levels. MRI revealed multiple plaques in the white matter of the brain and brainstem that did not accumulate gadolinium. Additionally, dry eyes were observed, and only after a biopsy was the patient diagnosed with Sjogren's syndrome (SS), although the course of the disease in this patient was very similar to chronic progressive MS and required a differential diagnosis. Unfortunately, an erroneous diagnosis of MS is made in 5-10% of cases [50].
SS and MS are chronic immune-mediated diseases with exacerbations and remissions, developing in young people, the pathogenesis of which is still not well known. These nosological forms have similar etiological, pathogenetic, clinical and neuroimaging indicators. They are characterized by a multifactorial etiology; autoimmune, inflammatory and neurodegenerative processes as the main pathogenetic mechanisms; marked variability in the clinical picture and often unpredictable prognosis.
Autoimmune mechanisms are of great importance in the pathogenesis of SS and MS (Table 1).
SS and MS are diseases that are associated with HLA class II genes of the major histocompatibility complex. Noteworthy is the presence of multiple associations with HLA genes in these diseases, which indicates, on the one hand, their multifactorial genesis, and on the other, their possible genetic heterogeneity. This locus is the only polymorphic genetic system whose association with autoimmune diseases has been convincingly demonstrated through many years of population studies [3, 7, 8, 10, 11, 18, 21, 27, 29, 30, 32, 34, 41, 44-46 , 54].
SS and MS are chronic inflammatory diseases, with MS characterized by a Th1 type of immune response, and SS by a Th1- and Th2-mediated type of immune response [2, 16, 22-25, 31, 40, 48] (Th1 - IL-2 , IL-3, lymphotoxin, IF-γ, GM-CSF, tumor necrosis factor α (TNF-α); Th2 - IL-4, IL-5, IL-6, IL-9, IL-10, IL-13 , IL-3, IF-γ, GM-CSF, TNF-α)[].
SS is a systemic autoimmune disease of unknown etiology, characterized by lymphoplasma cell infiltration of secreting epithelial glands with the most common lesions of the salivary and lacrimal glands [2, 28]. In patients with primary SS, various antibodies are detected in the serum. Neurological manifestations in SS are very often similar to the clinical picture of MS. In both diseases, patients may exhibit hypoesthesia, spasticity, hyperreflexia, ataxia, radicular disorders, hemiparesis or hemiplegia, dysarthria, cerebellar speech disorders, nystagmus, internuclear ophthalmoplegia, etc. Patients with SS with spinal cord lesions may develop lower paraparesis, tetraparesis, acute or subacute transverse myelopathy, chronic progressive myelopathy, neurogenic bladder, pelvic organ dysfunction, Brown-Séquard syndrome. If the optic nerve is involved in the pathological process in patients with SS, there may be monocular or binocular vision loss or optic neuritis/neuropathy. Some symptoms of central nervous system damage are presented in both MS and SS (Table 2).
In the majority of patients with SS, nonfocal neuropsychological disorders predominate [37]. Thus, according to J. Anaya et al. [6], when observing 120 patients with primary SS according to European criteria, in 3 of them, central nervous system damage was similar to other neurological diseases - MS, optic neuritis and complicated migraine. All these patients had elevated levels of anti-Ro antibodies. The literature [9] describes a case of the development of Devic's neuromyelitis optica in a patient with primary SS; in another observation [20], the patient had neuritis of the right optic nerve at the age of 10; in subsequent years, both optic nerves and the medulla oblongata were involved in the pathological process, and the diagnosis of SS was made later, when dry syndrome appeared. Similarities in the clinical picture were also noted by C. Iñiguez et al. [26], who among 100 patients with MS identified 2 female patients with xerophthalmia and xerostomia and positive Ro-antibodies. One of these women had recurrent episodes of left limb weakness and ataxia, and brain MRI showed hyperintense lesions in the periventricular white matter. The second patient diagnosed with MS (diagnosed at 53 years old) had multiple hyperintense foci on MRI of the brain and sicca syndrome.
MRI of the brain in patients with SS and CNS lesions may have a neuroimaging picture similar to MS (Table 3).
Approximately ⅔ of patients with SS with acute central nervous system damage are characterized by the presence of multiple areas of hyperintense signal on T2- and proton-weighted images of the brain [4, 5, 33].
B. Thong and N. Venketasubramanian [47] observed a 40-year-old patient with multiple neurological symptoms of the spinal cord and brain, in whom MRI of the brain and spinal cord during an exacerbation revealed a hyperintense signal and swelling of the spinal cord. Later, antinuclear antibodies and anti-Ro antibodies began to be detected in her, and then dry syndrome, which made it possible to diagnose SS. Also, the presence of brain damage according to MRI data has been shown [37] in neurologically asymptomatic patients with SS in the form of small lesions in the white matter of the brain.
In SS, the spinal cord may also be involved in the pathological process (Table 4), which is manifested by the development of transverse myelopathy, often recurrent or chronically progressive, dysfunction of the pelvic organs, and Brown-Séquard syndrome is less common.
Subacute or acute transverse myelopathy has also been noted as a consequence of inflammatory ischemic vasculopathy with the development of small vessel angiitis. J. de Seze et al. [15] among 11 patients with myelopathy against the background of SS, 7 had acute and 4 had chronic variants of the course, and in 7 cases optic neuritis was simultaneously present, which in 4 patients fully met the criteria for Devic’s syndrome. S. Delalande et al. [14], having conducted a retrospective study among 82 patients (65 women and 17 men) with primary SS, spinal cord damage was detected in 29 patients (acute myelopathy in 12, chronic myelopathy in 16 and motor neuron disease in 1 patient) and 13 patients had optic neuropathy.
Some authors believe that these antibodies represent the result of cross-reactions of anti-myelin antibodies or viral antigens with Ro/-SSA molecules and other autoantigens. If anti-Ro, antinuclear antibodies, anti-nDNA and anti-cardiolipin antibodies are detected in patients with MS, then these patients should be re-evaluated for rheumatic disease, which should be treated with immunosuppressive therapy (cytostatics, glucocorticosteroids).
In patients with SS, immunological examination reveals elevated titers of rheumatoid factor (RF), antibodies to soluble nuclear antigens Ro(SS-A), La(SS-B) in the serum, and antibodies to the Ro(SS-A) antigen are observed in 50% of patients, and antibodies to the La(SS-B) antigen - in 40-50% [43]. However, anti-Ro antibodies can be detected in 2-15% of cases in patients with MS (Table 5) [12].
Oligoclonal bands in the CSF are characteristic signs of MS; they are detected in 95% of cases and are included in the diagnostic criteria for this disease. However, these changes in the CSF can also be observed in SS (Table 6), but the differences are that patients with MS have numerous bands, while in SS there are only 1 or 2 bands.
A study of CSF in 17 patients with primary SS [19, 51] showed that intrathecal synthesis of oligoclonal IgG bands was detected in 6 of 8 patients with clinical manifestations of nervous system involvement and 5 of 9 patients without nervous system involvement. Intrathecal synthesis of antiviral IgG antibodies to measles, mumps, and herpes simplex viruses was observed in 8 of 17 patients with SS and 7 of 12 with MS. In the CSF, IgG antibodies to myelin were detected in 3 of 12 MS patients and in none with SS.
In a recent study conducted in Sweden using a free-sampling method, 30 patients with MS were recruited and tested for the presence of SS according to the Copenhagen criteria. 5 of these patients had keratoconjunctivitis sicca, 1 had xerostomia, and 3 had histologically confirmed sialadenitis with decreased saliva production, but none of these patients were diagnosed with SS [42]. In another study [52], among 12 patients with relapsing-remitting MS living in Asia, 6 individuals were identified that fully corresponded to the diagnosis of primary MS. At the same time, J. de Seze et al. [15], studying the frequency of SS in 60 patients with primary progressive MS according to clinical, laboratory, instrumental diagnostic criteria and biopsy results, revealed that 10 (16.6%) had 4 or more criteria for the diagnosis of SS. The detected frequency exceeds the general population (1-5%). In another study [39], when examining patients diagnosed with primary progressive MS for the presence of diagnostic criteria for SS using the European criteria for SS, the Schirme test, salivary gland scintigraphy, salivary gland biopsy and serological tests (antibodies Ro/SS-A, La/ SS-B and antinuclear) SS was diagnosed in 9 MS patients (at least 4 classification criteria). All of these patients were women with a mean age of 46.6 years at the onset of symptoms. Spastic paraparesis was observed in all of them, and MRI of the spinal cord revealed changes in the majority, but anti-Ro and anti-La antibodies were negative in most cases. Other authors [17, 35, 36] among patients with MS also identified both individual symptoms characteristic of SS and diagnosed SS as a nosological form, while in a large study conducted by J. Noseworthy et al. [38], among 192 patients with MS, not a single patient with clinical manifestations of SS was identified.
The issue of the influence of pathogenetic therapy on the course of the disease is currently being discussed in the literature [50]. Thus, L. de Santi et al. [13] observed a 48-year-old woman with relapsing-remitting MS, with a good response to interferon 1a-beta and dry syndrome that developed 29 years after the diagnosis of MS, which was confirmed instrumentally and according to biopsy.
Thus, in patients with SS and MS, similar data from clinical, neuroimaging and laboratory studies are often detected, which often creates difficulties in making a differential diagnosis between these nosological forms.
[]The following abbreviations are used: IL - interleukin, IF - interferon, Th - T-lymphocyte helpers, GM-CSF - granulocyte-macrophage colony-stimulating factor.
Multiple sclerosis
Quite often there are so-called. unexpressed forms in which some of the cardinal symptoms are absent. So, sometimes there is no nystagmus, no intentional tremor, no speech disorder, or these symptoms are very mild, and the whole picture of the disease is limited to one or two phenomena. Usually, however, the existing phenomena are sooner or later joined by other cardinal symptoms of the disease, but despite this, the diagnosis can sometimes still be difficult. So, for example, if nystagmus is present, especially if there is also a speech disorder and some sense of balance disorder at the same time, one can easily mistakenly assume Friedreich’s disease, especially since the latter may sometimes lack such characteristic phenomena as movement coordination disorder, scoliosis , clubfoot. Only a detailed anamnesis, a thorough analysis of the present phenomena and sometimes the further course of the disease can in such cases find out what is going on. Similarly, with hereditary cerebellar ataxia, some symptoms are observed that can exist as the only phenomena in mild multiple sclerosis: scanned speech, increased reflexes, onset of the disease after 20 years. Here, in the differential diagnosis, one must pay attention to the hereditary, family nature of the disease and the predominance of cerebellar phenomena. The other group consists of atypical forms due to the predominance of some unusual symptom. In this case, it is necessary to distinguish between two types of cases. In some cases, all or almost all of the cardinal symptoms of the disease are present, but in addition there is some other completely unusual phenomenon. Thus, sometimes there are cases of multiple sclerosis with muscular atrophy on the extremities, accompanied by a more or less pronounced degeneration reaction, or temporary disorders of the bladder (incontinence, urinary retention), or trophic disorders (hair loss, brittle nails, blistering rashes, etc.) . P.). The presence of these unusual phenomena can sometimes make one hesitate in the diagnosis, but the simultaneous existence of such symptoms as intentional tremors, nystagmus, scanned speech, changes in the fundus of the eye usually quickly eliminates doubt.
It is much more difficult to make a diagnosis in another category of cases, when only one unusual phenomenon is observed, while there are no cardinal symptoms of the disease, or there is only one of them. Thus, sometimes a patient suffers for a long time from attacks of dizziness, during which it seems to him that all the objects around him are spinning with very great speed and that he himself is participating in this rotational movement; sometimes there may also be subjective light sensations or darkening in the eyes and headache. For the most part, these phenomena subside when the main symptoms of multiple sclerosis are detected. It is clear how difficult it is, in the absence of a set of characteristic symptoms (intentional tremors, nystagmus, scanned speech, pallor of the optic nipple, gait disorder), to recognize the actual cause of an unusual phenomenon. Moreover, sometimes, although rarely, there may be a combination of unusual symptoms such as dizziness, headache and especially epileptiform seizures with fever, so that there may be an assumption of encephalitis or a brain tumor. Absence of nipple congestion, vomiting, confusion, etc. allows us to exclude such an assumption.
Of great interest are atypical forms that simulate some other organic disease of the nervous system. This category includes:
A form simulating amyotrophic lateral sclerosis, due to the presence of typical amyotrophic paralysis and bulbar phenomena. Distinctive features from Charcot's disease are a slower and fluctuating course and less pronounced muscle atrophy.
A form similar to transverse myelitis, or a paraplegic form, in which spastic paralysis of the lower extremities, increased knee reflexes and disorders of the bladder and rectum are observed. The difference is that with syphilitic transverse myelitis, disorders of sensitivity, bladder and rectum are more pronounced and none of the cardinal symptoms of multiple sclerosis are observed, even in the form of a hint.
The form simulating hemiplegia is very important and leads to the consideration of apoplectiform seizures that occur in multiple sclerosis. Apoplectiform attacks are sometimes observed at the beginning or during the course of the disease and can be repeated 1-2-3 or even 5-6 times. They differ from apoplectic attacks due to cerebral hemorrhage in that they are less severe and are accompanied by high temperature (this sign cannot be considered, of course, characteristic, since an increase in temperature can also be observed with apoplexy). The hemiplegia that mostly remains after them is usually temporary and can completely disappear. It affects only the limbs or both the limbs and the face, and may be accompanied by aphasia and paralysis of the eye muscles. Sometimes it is observed in the form of hemiplegia alternans. In some cases, hemiplegia may be due to concomitant hysteria. The diagnosis is not difficult to make if nystagmus and intention tremor exist simultaneously. Sometimes the hemiplegic form of multiple sclerosis can be confused with a brain tumor.
Very rarely there is a form simulating syringomyelia with hypertrophic cervical pachymeningitis, which can give rise to an erroneous diagnosis due to pain and especially paralysis with limb contracture. But, in contrast to syringomyelia, there is no characteristic sensitivity disorder, muscle atrophy, sclerosis and peculiar trophic disorders.
To put an end to forms that simulate other organic diseases of the nervous system, it is also necessary to point out the bulbar form of multiple sclerosis, in which disorders of phonation, swallowing and chewing, dizziness, attacks of difficulty breathing, sometimes glycosuria, tachycardia can give a picture very similar to progressive bulbar palsy . If there is also muscle atrophy, then one can mistakenly assume the bulbar form of amyotrophic lateral sclerosis. With the so-called In the cerebral form of multiple sclerosis, epileptiform seizures are observed, often alternating with apoplectiform seizures, a noticeable weakening of memory and mental abilities, sometimes delusions and hallucinations or bouts of causeless violent laughter (or violent crying). Such cases can sometimes simulate a brain tumor, but they can even more likely be confused with general progressive paralysis, especially if there is also a speech and gait disorder at the same time. When most of the cardinal symptoms of multiple sclerosis are present, the diagnosis is not difficult to make. In their absence, it must be remembered that speech disorder in progressive paralytics has characteristic features. Namely, it intensifies with emotional excitement and fatigue, in addition, the speech is not chanted, but stumbled, and later becomes completely illegible (the patient misses syllables in long words, rearranges them, etc.). In addition, trembling in progressive paralysis has a completely different character, and the disorder of mental abilities, physical and spiritual decline are much more pronounced.
Atypical forms are relatively rare due to an abnormal clinical course. This includes all those cases in which the disease begins with an apoplectiform or epileptiform attack in the midst of apparent complete health and then proceeds quickly, ending in death after 6-8 months. When a patient, as is sometimes observed, dies during such an initial apoplectiform attack, the diagnosis can, of course, be made only on the dissecting table.
Finally, there are also atypical forms due to a combination of multiple sclerosis with tabes dorsalis, progressive paralysis and especially hysteria. Cases of the latter kind are observed quite often and are important in that they can be mistaken for simple hysteria. Indeed, hysteria can simulate most of the cardinal symptoms of multiple sclerosis, so that it is sometimes difficult to decide whether we are dealing with hysteria under the guise of multiple sclerosis, or with a combination of both diseases. In addition to the history and the presence of hysterical stigmata (concentric narrowing of the visual field, anesthesia of the pharynx, hysterogenic areas, hemianesthesia, etc.), one must also take into account the features that the symptoms themselves represent. Thus, speech during hysteria is, in essence, not scanned, but rather incorrect, since some syllables are pronounced slowly, while others are pronounced quickly. Trembling also presents some differences from trembling in multiple sclerosis and, in particular, does not have a clearly defined intentional character. In the same way, reflexes represent characteristic features. It goes without saying that the observation of a pale optic nipple with an ophthalmoscope is decisive for the diagnosis, since this symptom does not occur in hysteria.