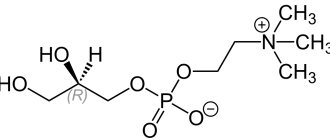
Excited brain
In antiquity, epilepsy was called a “sacred disease” sent by the gods. On the one hand, its manifestations were frightening, but on the other, people suffering from this disease often had outstanding abilities. In the modern understanding, epilepsy is a heterogeneous group of diseases, the clinical picture of which is characterized by repeated convulsive attacks (fits). They arise as a result of synchronous excitation of all neurons of a separate area of the cerebral cortex - the epileptogenic focus. Sometimes an epileptic attack is preceded by an aura, manifested in the form of unexplained lightheadedness, sound or visual disturbances, depending on the location of the epileptic focus. Seizures occur as brief, involuntary spasms in one part of the body (partial seizures) or throughout the body (generalized seizures). Generalized seizures are often accompanied by loss of consciousness.
Article on the topic What to do if you have epilepsy, and how to help during an attack? Advice from a scientist Seizures can happen less than once a year, or up to several times a day if the disease is severe. Often the causes cannot be determined, and then they talk about idiopathic epilepsy. An international group of scientists led by specialists from the University of Melbourne found that 12% of idiopathic epilepsy is caused by a mutation of the DEPDC5 gene (the protein encoded by this gene is involved in signal transmission within neurons).
Secondary, or symptomatic, epilepsy can have the following causes: brain damage in the prenatal or perinatal period (hypoxia or birth trauma), head trauma and stroke causing brain hypoxia, brain infection, such as meningitis and encephalitis, parasitic diseases, brain tumor. There is also so-called cryptogenic epilepsy. This diagnosis is made when the cause of generalized symptomatic epilepsy cannot be accurately determined from research results.
An epileptic seizure can be triggered by factors affecting brain activity: hormonal changes (in particular, menstruation), flickering light or flashing images (when riding a train, in a car), intoxication (alcohol, some medications, toxic substances). There are also equivalents of epileptic seizures in the form of sudden onset mood disorders, disorders of consciousness, and also, in the case of severe disease, characteristic changes in personality and intelligence.
Two diseases are directly related to the course of epilepsy: migraine and depression. Epidemiological studies have shown that every fourth patient with epilepsy suffers from migraine, and the incidence of epileptic seizures in people with migraine reaches 17% or higher. In this case, depression is detected in 20–55% of patients with constant seizures and in 5–10% of people with controlled seizures. This association is most often observed in patients with a partial form that is resistant to treatment.
Article on the topic
Expert: A cure for epilepsy can be achieved in 65-75% of patients. Often, depression in epilepsy is not diagnosed, but that is exactly what it is, according to a study conducted at the Department of Nervous Diseases, Faculty of Postgraduate Education, Perm State Medical University named after. I.M. Sechenov, is the most important factor influencing the patient’s quality of life (its influence is higher than that of epilepsy itself).
Treating epilepsy means preventing seizures
It is important to start treating the disease from the very beginning, then the likelihood of curing epilepsy increases significantly.
Most often, the prognosis for recovery depends on the form of the disease. As a rule, with genetic forms of epilepsy it is easier and faster to recover , the course is smoother, the amount and dose of anticonvulsants is smaller, the duration of treatment is shorter, patients do not have other symptoms of damage to the nervous system, do not lag behind their peers in development, and do not have pronounced changes in behavior. This does not apply to all forms of hereditary epilepsy, but to most.
There are forms, such as epileptic encephalopathies , in which delay or incorrectly selected treatment leads to rapid regression or underdevelopment of speech, mental retardation, and mental changes. Even with subsequent correctly selected anticonvulsant therapy, it is not possible to restore lost skills, speech, or improve behavior.
Some benign forms of epilepsy may not be treated.
And other, severe forms of epilepsy do not respond to multiple antiepileptic drugs; the outcome of the disease often depends on early onset and the correct choice of antiepileptic drug.
Curability has been proven
According to the European Commission on Epilepsy, this disease affects about 50 million people, or up to 1% of the world's population. In Russia, according to the Ministry of Health, epilepsy occurs with a frequency of 1.1 to 8.9 cases per 1000 people, but, according to experts, this disease is not sufficiently diagnosed. According to the chairman of the Russian Anti-Epileptic League, Professor Gagik Avakyan, in the vast majority of medical institutions epilepsy is detected using routine electroencephalography, the effectiveness of which is at best 30%. But to make an accurate differentiated diagnosis, at least a 12-24-hour video-EEG is required, capturing the time of night sleep (the effectiveness of the method is 88-95%). This procedure is usually available in paid medical institutions.
Few people know that 70% of children and adults newly diagnosed with epilepsy can be successfully treated (that is, seizures are completely controlled) with antiepileptic drugs. After two to five years of successful treatment, approximately 70% of children and 60% of adults can stop taking medications without risk of relapse. After this, if the patient follows the basic rules of the regime: sleeps for 7–8 hours, completely abstains from alcohol, the attacks, as a rule, do not return.
Article on the topic
“We are superfluous everywhere.” The story of a family raising a child with epilepsy Yet in developing countries, three quarters of people with epilepsy do not receive the treatment they need.
In Russia the situation is, of course, better, but different from that in developed countries. “Over the past 20 years, a lot of work has been done in pediatric epileptology, but at the same time, there is no reliable connection between pediatric and adult services,” says Professor Andrey Petrukhin, President of the Association of Epileptologists and Patients . “The adult neurological network is not ready to accept the patient and continue his treatment with an effective drug, which leads to loss of remission.”
Indeed, patients with epilepsy are observed either by neurologists or psychiatrists who are not able to understand the intricacies of diagnosis and treatment of this complex disease. Epileptologists in Russia are trained within the framework of postgraduate education programs, in particular at the Russian National Research Medical University named after N.I. Pirogov, but so far there are specialists not in all regions.
The problems also include the inaccessibility of drugs, and this is not only due to the high cost of the latter. “Unfortunately, new drugs are often unavailable to Russian patients due to the fact that they have not been registered in the Russian Federation,” says epileptologist, Ph.D. Yuri Shiryaev (University Headache Clinic) . “We do not have the right and opportunity to use effective medications recognized by the world medical community.”
Juvenile myoclonic epilepsy (JME) as a separate form of idiopathic generalized epilepsy with age-dependent onset was identified in 1969 by D. Janz [1], although there is evidence in an earlier work by this author, jointly with W. Christian, in which it was named “impulsive petit mal” [2]. In addition, there is the first description of the patient, which dates back to 1867 [3]. However, despite the long period of study of JME, there are still questions and unresolved problems regarding its diagnosis and treatment.
The article provides a comparative analysis of the results of our research in accordance with the works of other authors. The results of the study by V.A. were compared. Karlova and N.V. Freidkova [4] 72 patients with JME, as well as a new sample of patients over the past 5 years (20 people) with research materials from domestic and foreign authors relating to the same time period. The evolution of the problem in the aspects of clinical diagnosis, therapy, prognosis of the disease and social adaptation of patients with JME is considered.
The age of onset of this form of epilepsy in the literature is indicated with a wide range, which is associated with the unfixed point of onset of the disease. This is explained by the fact that the onset of the disease, as a rule, is myoclonus, to which patients, and often doctors, do not attach due importance. If in our previous studies an early onset was noted at 10 years of age, and a late onset at 26, now we have recorded a case of an earlier onset, namely at 9 years of age.
As for the genetic aspect of JME, in the works of F. Elmslic et al. [5] and later other authors [6-8] identified pathological genes - BRD 2
and
EFHCI
with a locus on chromosomes 6p21, 6p12-p11 (
EJM 1
) and 5q14 (
EJM2
) with a defect in one of the genes - myoclonin. In addition, the presence of widespread disorders at the microstructural level in the white matter of the brain was confirmed - in the frontal lobe and corpus callosum. The presence of these changes is associated with frontal cognitive dysfunction, which was noted by D. Janz [1], who was the first to describe this form of the disease. He also emphasized that, although most cases of JME are isolated (single cases), approximately 1/3 of patients have a family history of epilepsy. Data from P. Thomas et al. [9] indicate that in families of patients with JME, there is a segregation of patients with idiopathic generalized epilepsy, and approximately 5% of first-degree relatives have epilepsy.
According to our latest observations, epilepsy in the family was observed in 1 relative of the first degree and in 5 relatives of the first degree according to previously published studies. In 2014, a study [10] was conducted on clinical and genealogical data in probands with JME and their family members. We studied 13 unrelated families in which at least 2 members suffered from epileptic seizures. Neither maternal nor any other mode of inheritance was found. A heterogeneous phenotype was found in individuals of the second and third degrees of consanguinity: in families with JME, other forms of epilepsy are found, in particular with generalized tonic-clonic seizures (GTS) in individuals with late onset JME.
Here is our own observation.
An 18-year-old patient complained of seizures from the age of 11 - “the same as the mother’s” upon awakening, and a single generalized convulsive seizure (GSE) at the age of 13. For the last two years he has been taking Convulex 750 mg 2 times a day, there have been no attacks. The breakdown of clinical remission began with the occurrence of an attack “around sleep” (when falling asleep).
Born from a 23-year-old mother, labor was at term, induced. The patient's mother was previously treated with a diagnosis of JME: she dropped objects from her hands, and had morning twitches in her hands. As prescribed by a neurologist, she took Luminal for a year, and then phenytoin (diphenin) for three years, which naturally intensified the attacks. At the age of 20, she independently stopped taking prescribed antiepileptic drugs (AEDs), at the same time the attacks practically stopped: only twitchings provoked by psycho-emotional stress remained.
An examination of the patient's neurological status revealed mild Babinsky asynergia. MRI of the brain showed a large subarachnoid cyst in the left frontoparietal region, undoubtedly congenital. The previous EEG showed mild background disorganization, absence of photosensitivity and an epileptiform response to hyperventilation. At the same time, in the slow-wave sleep phase, epileptiform discharges of the acute-slow wave and spike-wave type were recorded, mainly arising in the frontal leads, local or sometimes generalizing. The latest EEG shows their significant reduction.
There are a number of features in this observation. Firstly, the mother of a patient with a clinical picture of JME at the age of 20, against the background of self-cancellation of AEDs (which, by the way, were contraindicated for her), only a rare psycho-emotional provocation of myoclonus remained. Secondly, in the daughter, the combination of morning myoclonus with isolated sleep-related generalized seizures (GSEs) provides a known basis for the diagnosis of JME. At the same time, we believe that this case can be qualified as an “erased” form of JME, since there were no characteristic shudders and falling of objects from the hands. It can be assumed that the mother had an “erased” form of JME. In principle, these observations support the existence of a problem of sleep-related myoclonus, perhaps outside of epilepsy.
As is known, JME has a characteristic clinical pattern: bilateral, often asymmetrical irregular arrhythmic jerk-like myoclonic twitching of the muscles of the shoulder girdle and the arms themselves of varying amplitude. At the same time, myoclonic twitches are largely ignored by patients, and therefore patients, as a rule, do not contact doctors with these complaints. However, they can hardly be called unconscious, since the doctor’s leading questions are followed by a description of these attacks to the patient. Rather, this phenomenon can be explained by the characteristic features of the psyche of patients (anosognosia) [1].
Another characteristic clinical manifestation of this form of epilepsy is a generalized myoclonic phenomenon - startle. Unlike physiological shudders, this manifestation of the disease in JME occurs not so much during sleep as during the period of awakening. In one of the patients we observed, shudders predominated when going to bed. This brings JME closer to awakening GTCS epilepsy, in which seizures can occur around sleep.
The combination of myoclonus in most cases with rare GTCS (60%) is confirmed. According to our latest observations, the average time for the onset of GTCS after the onset of myoclonus was about 4 years. The combination of myoclonus only with absence seizures remains a rather rare phenomenon: we previously observed only one case, over the past 5 years there have been no such cases in our observations.
The combination of myoclonus, GTCS, atypical absence and aura, which causes some diagnostic difficulties, is reflected in the following clinical observation.
Patient X.
, 29 years old, applied in July 2013. She is a psychologist by profession, but does not work.
The prenatal and postnatal anamnesis is unremarkable, heredity is not burdened, according to the mother, it was “squeezed out” during childbirth. Against the background of apparent health, but at the same time chronic lack of sleep, in April 2012, during a trip to work on a train, the first and only GTCS occurred with a preceding feeling of the unreality of what was happening (the patient could not describe it more accurately). By the time he contacted us, therapy had already been prescribed - Topamax 50 mg per day, Lamictal 100 mg 2 times a day. This caused a deterioration - the appearance of shudders, more often associated with sleep.
MRI on one of the axial basal slices in T2 mode revealed increased signal from the pole of the left temporal lobe. Sleep EEG is represented by episodes of generalized spikes and polyspike waves, single and/or extended to several seconds (in these cases, reminiscent of atypical absence seizure).
Considering that the patient had a history of surgery for bilateral polycystic ovary syndrome (the menstrual cycle is not disrupted), and the first-line drug of choice for the dominant clinical manifestation of myoclonus may be levetiracetam, it was decided to cancel Lamictal and add Keppra 500 mg 2 times a day. In September 2013, there was a second GTCS, which the patient amnesiced, but at the same time, apparently, she felt its beginning (she left the kitchen and went to bed). In addition, there is an isolated, difficult-to-identify state of derealization. The twitching decreased. The dose of Keppra was increased to 750 mg 2 times a day and Topamax 50 mg 2 times a day was added (taking into account the patient’s excess body weight). There followed an increase in the frequency of attacks up to one per week while awake, and the patient independently discontinued the AED. After which there was no GTCS, however, general tremors occurred during sleep at night. During 12-hour EEG monitoring, bilateral synchronous peak-wave and polypeak-wave discharges with a frequency of 3.5-4 and a duration of 3-5 s were recorded during periods of wakefulness and sleep.
This case initially presented diagnostic difficulties: the differential diagnosis was made between JME and phenocopy JME. Uncharacteristic, although possible, for JME was the onset of the disease with GSP, an increase in convulsive seizures during the course of the disease, and an EEG pattern largely represented by a correlate of atypical absence. Another feature that initially led to discussion of the diagnosis of JME was the appearance of myoclonus, usually associated with sleep, due to its appearance after the prescription of a combination of Topamax (prescribed due to the patient’s excess weight) with lamotrigine. Since both of these drugs, although rarely, can provoke the appearance of myoclonus, the mere fact that myoclonus occurred not after waking up, but during sleep, did not negate the diagnosis of JME. Finally, one more significant circumstance: the self-cancellation of the drugs coincided with the spontaneous remission of GSP, although the twitching persisted. Thus, this case demonstrates that myoclonus is the essence of the disease. As for GSP, it is obvious that the first of them was provoked by lack of sleep, and the subsequent appearance and increase in frequency of attacks must be regarded as the result of an ineffective or even paradoxical effect of AEDs.
Let's return to the diagnosis. The onset of the disease is from the age of 10, when short-term episodes of “freezing” and flinching were noted; the first attack that occurred on the train was apparently associated with sleep deficiency, and the generalized changes revealed by the EEG could indicate the idiopathic nature of the disease: JME refers to idiopathic epilepsy with a varying phenotype. At the same time, a patient with GSP has a difficult-to-identify aura, which also occurs in the form of an isolated manifestation; transformation of convulsive seizures from asynchronous to seizures of wakefulness are signs characteristic of symptomatic and, to a large extent, cryptogenic epilepsy. Already in the 1989 classification, a form of epilepsy was identified with syndromes that have signs of both focal and generalized. In the later proposed and still not accepted classification in the section “Epileptic syndromes and related conditions”, the above heading was removed [11, 12]. However, the new composition of the International League Against Epilepsy (IALE) Commission on the Classification and Terminology of Epilepsy, headed by I. Scheffer [13], recently proposed the introduction of a section “Unclassified epilepsies”, which includes cases not recognized as known electroclinical syndromes, or of unknown etiology. Thus, this case does not relate to specific electroclinical syndromes and can be classified as an unclassified form of epilepsy.
Since JME can initially manifest itself for a long time only as myoclonus, EEG is of particular value. EEG in JME is characterized by such features as the presence of short bursts of polyspike and spike waves of 3-6 v s (Fig. 1); discharges of generalized symmetrical spike waves 3 in s and rarely 1-2 in s; epileptic discharges characterized by high, sometimes gigantic amplitude. Frontal-lobar activity is detected more often (Fig. 2 and 3), sleep sharply activates epileptiform activity, while emphasis can also be detected in some other leads. Although clinically absence seizures were observed in isolated cases, during EEG recording, a correlate of absence seizures was recorded in 18.1% of early studies and in 15% of later studies, while an EEG correlate of myoclonus was recorded in half of the patients.
Rice. 1. EEG of patient Kh., 29 years old, with JME.
Rice. 2. EEG of patient X., 29 years old. Frontal lobar activity.
Rice. 3. Localization of the epileptic focus according to EEG leads (explanation in the text).
Focal EEG manifestations are widespread in patients with JME [14]. Thus, in the studies of E. Montalenti et al. [15], K.Yu. Mukhina et al. [16] revealed a high frequency of regional changes and asymmetry of diffuse peak-wave discharges. According to these authors, these “atypical” changes in the EEG are often the cause of an erroneous diagnosis of focal epilepsy with the phenomenon of secondary bilateral synchronization. In this regard, it is appropriate to mention that domestic authors emphasize: the classic clinical picture is fundamental in making a diagnosis, and EEG is just an auxiliary additional research method.
Unfortunately, despite the characteristic clinical picture of the disease and progress in research methods, the problem of diagnosing and prescribing correct therapy still remains open, although positive trends are observed. If previously a correct diagnosis was made and adequate therapy was prescribed only to 34.8% of patients, then according to recent studies, initial therapy was correctly selected for 50%. We note, by the way, that phenytoin is still sometimes prescribed.
The diagnostic problem is confirmed by some foreign works. Thus, a retrospective observation of 200 patients with JME who underwent outpatient examination in 2014-2015. [17], found an incorrect diagnosis in 49 cases with the onset of the disease with GTCS and registration of generalized spike-wave or polyspike-wave discharges on the EEG in 56% of cases. In cases where the disease began with myoclonic seizures, the diagnosis did not cause difficulties. Almost half of the patients were prescribed inappropriate AEDs; the rest were recommended to be monitored. Compared to the 1998 study, the rate of misdiagnosis has become lower and the time to correct diagnosis is shorter. However, diagnosis at a glance still remains a challenge among neurologists, even when typical EEG changes occur.
A substantiated diagnosis of JME, however, does not guarantee an optimal response to therapy. This fact was reflected in the work [18] on a study of 116 patients with JME, observed for at least 18 months each. 68 patients had no seizures over the past 12 months, and 48 had at least one seizure of a different type. The most common negative factors found in the latter group were: short follow-up period, medication other than valproic acid (VA), poor adherence to therapy. In particular, in this regard, the authors recommend wider use of valproate. Other researchers [19] noted that 19% of 201 patients with JME with no response to VC showed significant correlations and associations with mental disorders. This, as well as other treatment problems and causes of relapse are discussed in this article.
One of the problems in the treatment of JME, as with other forms of epilepsy, is adverse reactions.
Patient Z.
, fell ill at the age of 11-12, when twitching appeared in the morning after sleep, she dropped objects from her hands. MRI and EEG studies were carried out, after which the disorders were regarded as functional tics. At the age of 15, she suffered from salmonellosis, against the background of which a sharp headache developed, accompanied by vomiting, aggravated by verticalization. She underwent a course of treatment in the infectious diseases department, but the headache was not relieved, and a space-occupying process in the brain was suspected. In May of the same year, she was examined at the Scientific Center for Neurology, diagnosed with myoclonus and prescribed clonazepam and finlepsin. A positive effect of initial therapy was noted, but epileptiform activity remained on control EEGs. A year later, against the background of forced self-discontinuation of medications (the AEDs ended while on vacation), GTCS developed in the morning. The dosage of clonazepam was increased. Four years later, during the morning toilet in the bathroom, there was an episode of short-term loss of consciousness; there was no post-attack sleep.
At this time, the patient first contacted the authors of the article, and the treatment was changed: Depakine Enterik 300 mg 3 times a day was prescribed. The patient's condition improved significantly: no twitching was observed, and all subsequent EEGs were positive. However, adverse reactions appeared: weight gain, menstrual irregularities, hirsutism. It was decided to prescribe Keppra 750 mg 2 times a day and reduce the dosage of Depakine. Since the side effects of Depakine persisted, it was necessary to completely remove Depakine within a year, but after 3 months the EEG deterioration continued. When the Keppra dosage was increased to 2000 mg per day, the condition remained the same. During one of the attacks, the patient almost fell on the stairs; Depakine 300 mg/day was again introduced into therapy. A year later, a pronounced deterioration was recorded on the EEG; in agreement with the patient, it was decided to switch to combination therapy with Topamax 100 mg 2 times a day and Depakine Enteric 300 mg 2 times a day. The EEG began to show positive dynamics. Later, an attempt was made to remove Depakine, but the weakness in the arms increased, uncertainty appeared in the morning, eyes rolled, and negative dynamics were recorded on the EEG. It was decided to return to the dosage of Depakine 300 mg 2 times a day.
We present this case not to illustrate the side effects of VK, they are well known, but to show what difficulties can arise if it is intolerant. Perhaps we should have tried other forms of VC.
It is known that JME is more common in females in a ratio of 2:1. This reflects another side of the problem - the gender aspect. Despite the fact that valproates are effective first-line drugs for JME, in many cases they cannot be used in an effective dose in females of reproductive age due to severe side effects, as illustrated above. This problem was discussed by the European Academy of Neurology, and a recommendation was made to limit the use of valproate in this category of patients [20].
V. Puri et al. [21] tried to find the neurophysiological mechanism of the prevalence of morbidity in women using the method of transcortical magnetic stimulation in previously untreated patients. Increased activity was found in the cortical inhibitory neural network, “apparently due to a long period of cortical silence.” This phenomenon was found only in female patients, which the authors explain the increased sensitivity to this disease in the female cohort and, accordingly, the possible significance of the hormonal factor (estrogen).
In connection with the above, it is relevant to further search for an alternative drug VK for the treatment of JME in women.
Levetiracetam, which is highly effective against both myoclonus [22, 23] and in blocking interictal epileptiform discharges and photoparoxysmal response to EEG [24], can be a replacement for valproates in the treatment of JME.
In 2007, a study [25] was conducted on 30 patients treated with levetiracetam, of whom 80% achieved complete remission with levetiracetam monotherapy. In this case, the final therapeutic dosages ranged from 12 to 50 mg/kg per day. This study supports the possibility of considering levetiracetam as a first-line treatment for JME.
One of the authors of this article together with N.V. Freidkova [26] conducted a study that assessed the effectiveness of low doses of valproate and levetiracetam in the treatment of idiopathic forms of epilepsy; 13 of 23 patients had JME. At the same time, the group of patients with JME (both in combination with GTCS and without them) was the most representative in achieving the best therapeutic effect. Thus, complex therapy with the mentioned AEDs led to drug remission in 6 out of 8 patients; in 1 observation, the frequency of attacks decreased by more than 75%, and in another 1 - by more than 50%.
In another study [27] of the effectiveness of levetiracetam in monotherapy in 4 patients with JME (2 patients of whom were prescribed the drug for the first time in connection with pregnancy planning, the rest were transferred to carbamazepine therapy due to clarification of the diagnosis), the use of AEDs made it possible to achieve remission of attacks within 6 months
At the same time, according to some literature data [28], levetiracetam is not inferior in effectiveness to valproate. Domestic authors conducted an extensive retrospective study of a database of 1159 patients in the Volgograd region, of which 78 suffered from JME. The follow-up period ranged from 1 to 6 years. In 56% of cases, patients diagnosed with JME were on valproate monotherapy, in 17% of cases levetiracetam was used. The rate of achieving drug remission was 83%, while when using monotherapy with valproate - 92%, levetiracetam - 87%. Complete clinical EEG remission was achieved in 41% of cases, mainly with the use of valproate.
Also, along with the study of valproate, a prospective randomized study [29] was conducted on the use of lamotrigine in patients with the onset of JME in adulthood, which compared its effectiveness and tolerability relative to V.C. There is evidence that lamotrigine is effective and better tolerated in adult patients with JME, although the prevalence of idiosyncratic reactions may be a cause for concern.
It should be noted that modern scientific literature does not raise the question of the condition of children born to mothers with JME, as well as the role of male heredity. Let us give several examples from our practice, in particular in cases such as the one described below.
Patient L.
, 37 years old, first applied at the age of 18 after GSP, due to the fact that her menstrual cycle stopped (the attack was on the eve of the start of menstruation). The second GSP of sleep happened a week later, already against the background of menstruation. Subsequently there was only a single SHG. As for myoclonus, they recurred periodically. The patient complained of frequent disturbing dreams, for which reason phenazepam was prescribed. For the first time, when examining the patient, the EEG revealed acute-slow wave complexes of frontotemporal localization of an alternating type; the last EEG in a state of wakefulness showed an episode of generalized high-amplitude spike waves with maximum severity in the anterior parts of the brain. During all this time, the patient was pregnant three times, two pregnancies were terminated for social reasons, and one took place. As of January 2015, the child is 15 years old. He was distinguished by deviant behavior, a decrease in the level of intelligence; Until the age of 13, he had nocturnal and daytime enuresis, and studies in a special school.
Since the onset of the disease, the patient has been constantly taking Depakine Chrono 300 mg 2 times a day and lamotrigine (Lamolep) 100 mg 2 times a day. There are no convulsive attacks, but still shuddering in the morning.
It should be noted that the results of the study by K. Meador et al. [30] demonstrated a decrease in IQ (intelligence quotient) in school-age children whose mothers used VC during pregnancy by 7–10 points compared to other AEDs.
To date, a number of studies have been conducted on the long-term outcomes of using VC during pregnancy, the results of which indicate a 3-fold increase in the frequency of autism spectrum disorders and a 4-fold increase in autism in children, along with population indicators. Some studies give reason to believe that such children develop attention deficit hyperactivity disorder [31-33].
The effect of VK on the mental and physical development of children when used during pregnancy depends on the dosage, although the dose threshold has not been determined. This problem and a number of issues of pregnancy in JME against the background of VK monotherapy are discussed in detail in the article by P.N. Vlasov [34].
The presence of these problems is confirmed by the existence of cases such as the one given below.
Seizures occurred in a 6-year-old child born to a father with JME. The onset of the disease in my father began with a complex partial seizure in his sleep (automatism). He was prescribed initial therapy with carbamazepine (Finlepsin) 200 mg 2 times a day. Against this background, the patient, along with repeated complex partial seizures, developed attacks of “shuddering.” EEG showed regional and generalized spike and polyspike wave activity. Then, carbamazepine was gradually replaced by depakine 500 mg 2 times a day. Epileptic activity on the EEG regressed, and the twitching stopped. Two years later, taking into account clinical EEG remission, an attempt was made to reduce the dose of depakine to 600 mg per day with a drug concentration in the blood of 48.3-50.8 mcg/ml, however, absence activity appeared on the EEG during night sleep. When the dose was increased to 300 mg in the morning and 500 mg in the evening, there was a resumption of tremor in the hands. Complete remission was achieved with a dose of 1000 mg per day (500 mg per 2-fold dose). As for the child, the perinatal history, according to the parents, is not burdened, but it is observed by a neurologist with a diagnosis of “cerebral palsy, symptomatic epilepsy with rare convulsive seizures against the background of antiepileptic therapy.”
Unfortunately, genetic analysis was not performed in the described case, so the possible contribution of a genetic factor remains unproven. Another important, but unresolved issue for both the patient and his family members remains the issue of further prognosis of the disease.
In the work of J. Gaithner et al. [35], based on an analysis of patients observed for at least 25 years, unfavorable prognosis factors were identified: onset with GSP preceding myoclonus, failure of therapy and polytherapy. Contrary to conventional wisdom, long-term treatment is not necessary, except for those patients who have these unfavorable prognosis factors. At the same time, in our studies of 30 patients, discontinuation of AEDs after a 5-year drug remission was possible only in 9; in the remaining cases, a relapse occurred. It is possible that this phenomenon is to some extent associated with the late start of adequately selected therapy. According to our observations, over the past 5 years, there have been 3 cases out of 20 with complete clinical, drug and EEG remission during treatment with VK and/or levetiracetam.
In the study by K.Yu. Mukhina et al. [16] in 106 patients with JME, clinical remission was achieved in most cases (89.6%), and clinical EEG - in 22%. The majority of patients (79%) used monotherapy, the most frequently used was VK (56%), and less commonly, levetiracetam (13%). Unfortunately, despite the high effectiveness of treatment, the authors note a high percentage (92%) of relapses, most often at the stage of dose reduction by more than 50% and during the first year after discontinuation. The authors attribute this to such reliable factors as lack of sleep (23.5%), self-cessation, dose reduction or skipping AEDs, and replacement with generics (21%).
Difficulties in social adaptation, unusual lifestyle and poor cooperation with doctors in patients with JME were first mentioned by D. Janz and W. Cristian [2]. Similar changes, reaching the level of mental disorders, were registered in 25.6% [36, 37].
Since the peculiar mental disorders in patients with JME are associated with cognitive impairment due to the frontal lobes [20, 38], Brazilian authors undertook a special study in this direction on a contingent of 42 patients with an undoubted diagnosis of JME and 42 individuals in the control group using a battery of tests. The data obtained showed that patients with JME have worse adaptation in 2 significant aspects of life - work and family relationships, however, this factor is not correlated with cognitive impairment, but with a high frequency of attacks and impulsivity [39].
A significant factor for the patient and his relatives is the early prognosis of the disease. The authors of one of the studies [40] studied sensitivity to eye closure in JME and the effectiveness of prognosis in 76 patients with a minimum follow-up period of one year. 15.8% had a poor prognosis due to resistance to adequate AEDs, 19.7% were pseudo-resistant (i.e., mistreated), and 64.5% had a good prognosis. They also studied sensitivity to eye closure. It was found only in 4 (5.3%) patients and only in those with a poor prognosis. Thus, photosensitivity is likely to be a factor that worsens prognosis.
There are data in the literature [41] regarding the relationship between the photoparoxysmal response to EEG and optical coherence tomography data. In a group of patients with a photoparoxysmal response to EEG, an increase in the thickness of the retina nerve fiber layer
both eyes and the choroid of the right eye. The retinal thickness of both eyes was significantly less. The authors believe that these microstructural features may be associated with photosensitivity in patients with JME. Other studies [40] have shown that eye closure sensitivity (ECS) in patients with JME is a rare EEG finding and cannot be a reliable marker of poor prognosis.
It is known that a violation of cortical plasticity plays one of the main roles in the pathogenesis of JME. A group of Italian scientists [42] studied synaptic plasticity of the motor cortex using the method of paired associative stimulation in patients with JME and identified corresponding changes.
The data presented indicate positive changes in the diagnosis and treatment of JME. However, despite the characteristic clinical picture of the disease and the progress of modern research methods, as well as extensive clinical experience, the issue of diagnosing this form of epilepsy remains a problem for neurologists. Factors that aggravate the prognosis have been identified: a combination of myoclonus with GTCS, the presence of mental disorders, photosensitivity, lack of proper response to adequate AEDs. At the moment, the gender aspect of the disease still remains open, especially in relation to therapy. It is also necessary to accumulate scientific information about children born to mothers with JME and the role of paternal heredity.
The authors declare no conflict of interest.
Absolute evil
However, in recent years, specialists have encountered an unexpected problem associated with the government’s attempt to save on treatment at any cost. Expensive but effective original drugs are being replaced by cheap generics, which win tenders for preferential provision of drugs. “In epileptology there is a golden rule, recognized as an international standard of treatment,” says Yuri Shiryaev. “You cannot replace a drug that has achieved remission.” Russian studies fully confirm this rule.
Article on the topic
Gagik Avakyan: “Those suffering from epilepsy are often talented” For example, according to a professor at the Moscow Regional Research Clinical Institute. Vladimirsky Irina Rudakova, after switching from the branded form to generic analogues of topiramate within a year, loss of remission occurred in 75.6% of patients, status epilepticus - in 3.75%, emergency care or hospitalization was required in 51.9%. A switchback to the original drug was made in 86.2% of patients, after which the initial doses of Topamax were increased in 58.0%, the transition from monotherapy to polytherapy was made in 60.0%, and the initial level of seizure control was achieved only in 32.9% sick.
At the same time, the state does not gain anything: failure of remission in epilepsy is much more expensive, especially if we take into account not only direct, but also indirect costs associated with disability. In accordance with the Recommendations of the Expert Council of the Russian Antiepileptic League, it is preferable to start therapy with original drugs or with generics made according to GMP standards. In each case, continuous provision of the patient with the antiepileptic drug prescribed by the doctor must be guaranteed. In a patient in remission, any replacement of the drug (original with generic, generic with generic, and generic with original) should be avoided.
Discrimination based on illness
Laws prohibiting social discrediting of people with epilepsy were adopted in the USA only in 1990, in the UK and Australia in 1992. Until now, in China and India, epilepsy is considered an obstacle to marriage. In Russia, if you have been diagnosed with epilepsy at any time in your life, it is a contraindication for working as a doctor or nurse directly involved in treating patients, as a teacher, or as an artist. In Russia, Poland and Japan, persons who have ever been diagnosed with epilepsy are permanently deprived of the right to drive. In more liberal countries, the ability to drive is determined by the current absence of seizures and mental disorders.
Question answer
Epilepsy attack: how to provide first aid? Infographics The duration of the period from the last attack, allowing the patient to raise the question of obtaining a driving license, is, according to the recommendations of the International Bureau of Epilepsy, 2 years.
Diagnosis of epilepsy in
Diagnostics allows you to accurately establish a diagnosis and identify dysfunction of the brain.
For patients with epilepsy we provide:
- Electroencephalography (EEG) – registration of the total impulse activity of the brain using electrodes (leads from the scalp). The study gives doctors a clear idea of the location of the lesion in the brain;
- Long-term video-EEG monitoring (day, night, daily, multi-day).
- Interpretation of MRI images
- Neuropsychological diagnostics
Patients are managed by neurologists-epileptologists, who are among the best specialists in this field. Neuropsychologists and other specialists are involved in diagnosing patients with epilepsy.
Not just pills
In the fight against epilepsy, especially resistant forms, doctors use not only chemotherapy. According to Yuri Shiryaev, in pediatric practice and in adults with partial seizures, electrical stimulation of the vagus nerve is used with sufficient effectiveness.
The pulses are created by a generator installed under the skin (under the left collarbone or near the armpit). This operation refers to high-tech medical care. According to the doctor, a ketogenic diet is effective in children, and therapeutic plasmapheresis is used as an additional method. It is believed that it may enhance the body's response to medications.
In the USA, data have appeared on the effectiveness of electrical stimulation of the trigeminal nerve (in adults).
Treatment of epilepsy
Examination and further treatment of patients with epilepsy under the supervision of experienced epileptologists is carried out according to a well-established protocol. The main observation takes place on an outpatient basis in our medical center. If necessary, the patient may be hospitalized, for example, for some tests or if the patient's condition is serious.
Patients seen in our clinic can contact us any day if they have acute problems during their appointment. And get highly qualified help from an epileptologist, psychiatrist, psychologist, neuropsychologist.
In cases of focally caused epilepsy with a pharmacoresistant course, after a special preoperative examination of the patient, presurgical diagnosis can be performed.