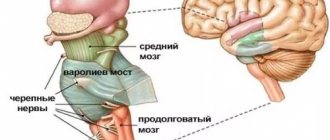
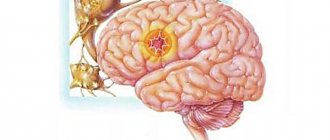
Myoclonic epilepsy is a type of epileptic seizure. Characterized by a milder flow. The disease first appears in infants or young children. The onset of myoclonic epilepsy in adulthood is atypical. This form of the disease is characterized by flaccid muscle twitching. Symptoms resemble tics or hyperkinesis. In some cases, a severe course is possible. In the international classification of diseases ICD-10, myoclonic epilepsy is coded G40.3.
You can undergo diagnosis and treatment of the disease in Moscow at the Yusupov Hospital. The examination is carried out by experienced neurologists and epileptologists using modern medical equipment. The therapy meets European standards of quality and safety.
Causes of myoclonic epilepsy
At present, the exact causes of the development of myoclonic epilepsy have not been established. However, doctors identify several predisposing factors. Among them:
- Hereditary predisposition. Certain types of myoclonic epilepsy are hereditary. For example, Unferricht-Lundborg disease, Dravet syndrome. If one of your relatives has been diagnosed with epilepsy, then subsequent generations are at risk. The probability of the disease occurring is 20-30%.
- Intrauterine infection. Some infectious agents are able to penetrate the placental barrier. As a result, the risk of developing not only myoclonic epilepsy, but also mental disorders and developmental defects increases. Myoclonic epilepsy develops at the end of the 2nd-3rd trimester.
- Diseases during pregnancy are not infectious. This group of diseases includes diabetes mellitus, thyroid pathology, kidney, liver, and heart failure.
- Uncontrolled use of drugs during pregnancy. Some drugs have pronounced teratogenic properties. Therefore, their intake negatively affects the development of the fetus. Doctors advise avoiding taking medications during pregnancy. If necessary, all prescriptions must be carried out by a doctor.
- Spontaneous mutations. The reasons for such changes have not yet been studied. Provoking factors are identified, the presence of which contributes to mutation processes. These include stress, sudden changes in temperature, and excessive physical activity.
SUB-SYNDROMES OF JUVENILE MYOCLONIC EPILEPSY.
Belyaev O.V.(1,2), Volkova E.A.(1), Vlasov P.N.(3),
- Medical Center for Neurology, Diagnostics and Treatment of Epilepsy “EpiCenter”, Volgograd
- State Budgetary Educational Institution of Higher Professional Education "Volgograd State Medical University" of the Ministry of Health of the Russian Federation.
- State Budgetary Educational Institution of Higher Professional Education "Moscow State Medical and Dental University named after A.I. Evdokimov" Ministry of Health of the Russian Federation
Juvenile myoclonic epilepsy (JME) (Jantz syndrome) is a common epileptic syndrome, accounting for about a third of cases with onset in adolescence. According to Wolf and V.A. Karlova [1, 2], the true frequency of JME in the population of patients with epilepsy is much higher than that indicated by most authors, but the disease is relatively rarely correctly diagnosed.
The criterion for the diagnosis of JME is the presence of massive myoclonic paroxysms (MPs) in patients, but they rarely occur in isolation. Previously, three types of seizures were considered characteristic of Janz syndrome.[3] Back in 1957, D. Janz and Christian described that the main clinical manifestations of this syndrome are MP, generalized tonic-clonic seizures (GTS) and absence seizures. Subsequently, with a more thorough study of patients with JME, publications gradually appear that describe the variability of the course of the syndrome. Thus, K.Yu. Mukhin and A.S. Petrukhin noted in 2000 that only 20–50% of patients experience short absence seizures of the juvenile type. And in isolated cases, pycnoleptic absence seizures that occur in childhood can transform into JME in adolescence, and predictors of transformation have been identified. “A pronounced myoclonic component in the structure of absence seizures (also the syndrome of epileptic myoclonus of the eyelids with absences), the presence of photosensitivity and initial resistance to valproate monotherapy in medium doses are the main predictors of the transformation of childhood absence epilepsy (CAE) or juvenile absence epilepsy (JAE) into JME.” [4]
And already in 2001, the committee of the International League Against Epilepsy proposed combining such forms as JME, JAE and epilepsy with GTCS into the group of idiopathic generalized epilepsies (IGE) with a variable phenotype. [5]
One of the first studies to identify JME phenotypes/subsyndromes was conducted by Martinez-Juarez in 2006 (n=257 patients with JME). In this article, the authors, according to the results obtained, presented 4 phenotypes or subsyndromes [6]:
- classic JME (72%)
- DAE transforming into JME (18%)
- JME with absence seizures in adolescence (7%)
- JME with astatic attacks (3%).
The authors also note that in families where Janz syndrome initially manifested itself in the classical form, subsequent generations are more likely to display the classical phenotype. But in families where JME was transformed from DAE, various forms were subsequently recorded.
Large population-based studies in Nova Scotia (n=682, follow-up 16 years) also showed the presence of four variants of the development of JME [7].
But a team of authors from Austria, after a retrospective study of patients with Janz syndrome (n=242, follow-up period 31 years), grouped patients by type of attack [8].
The purpose of our study was to analyze a sample of patients with Janz syndrome for the presence of phenotypes, as well as to assess the variability in the occurrence of different types of seizures and their combinations.
Materials and methods
The study was a retrospective clinical study using a database of 79 patients diagnosed with JME. During the work, a statistical analysis and an expert method for assessing the clinical picture of the examined group were carried out. Statistical processing was carried out in Excel (reliability of results p = 95%).
The results obtained are presented in the form of tables.
Table 1 Subsyndromes of JME
| Subsyndromes | % | N=79 |
| Classic version | 85,89 | 67 |
| JME transforming from DAE | 3,84 | 3 |
| YuME transforming from YuAE | 7,69 | 5 |
| JME with astatic attacks | 3,84 | 3 |
| Table 2 Occurrence of different types of seizures. | ||
| Myoclonus | 100% | 78 of 78 |
| Generalized TCH | 88,46 | 69 of 78 |
| Absence seizures | 16,67 | 13 of 78 |
| Astatic attacks | 3,84 | 3 of 78 |
Table 3 Features of clinical manifestations, depending on the subsyndrome.
| Types of attacks n = 78 | Classic JME | YME from DAE | JME with absence seizures | JME with astatic attacks |
| MP only | 5 | |||
| MP + Abs | 1 | 2 | ||
| MP + State Customs Committee | 60 | |||
| MP + Abs + GTK | 1 | 3 | 3 | |
| MP + Ac + GTK | 2 | |||
| MP + Abs + Ac + GTK | 1 |
MP - myoclonic seizures; Abs- absence seizures; GTC - generalized tonic-clonic seizure; As - astatic attack.
Conclusions.
The clinical picture in Janz syndrome is diverse and variable. In the Volgograd region, according to EpiCentre LLC, all 4 subsyndromes were identified, with the greatest degree of representation of the classical phenotype (86%).
It was determined that within the subsyndrome, the clinic can also be different, depending on the type of combination of different types of attacks.
Literature.
- Wolf P. Juvenile myoclonic epilepsy // In: Epileptic syndromes in infancy, childhood and adolescence / Eds. J. Roger, M. Bureau, Ch. Dravet et al. - Paris, 1992. - p. 316-327.
- V. A. Karlov Epilepsy in children and adult women and men: A guide for doctors. – M.: OJSC “Publishing House “Medicine” 2010. – 720 pp.: 244-248.
- Mironov M.B. Risk factors and relapse rates in patients with juvenile forms of idiopathic generalized epilepsy. dis. .cand. honey. Sci. M., 2005. 119 p.
- K.Yu.Mukhin, A.S.Petrukhin Idiopathic forms of epilepsy: taxonomy, diagnosis, therapy M.: “Art-Business Center”, 2000. - 319 p.
- Engel J.; International League Against Epilepsy (ILAE). A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001;42(6):796–803.
- Martinez-Juarez, IE, Alonso, ME, Medina, MT, Du Ron, RM, Bailey, JN, Lopez-Ruiz, M., Ramos-Ramirez, R., Leon, L., Pineda, G., Castroviejo, IP , Silva, R., Mija, L., Perez-Gosiengfiao, K., Machado-Salas, J., Delgado-Escueta, A.V., 2006. Juvenile myoclonic epilepsy subsyndromes: family studies and longterm follow-up. Brain 129, 1269–1280.
- Carol S. Camfield, Peter R. Camfield 2009 — Juvenile myoclonic epilepsy 25 years after seizure onset- Neurology September 29, 2009 vol. 73 no. 13 1041-1045
- Ulia Höfler, Iris Unterberger, Judith Dobesberger, Giorgi Kuchukhidze, Gerald Walserb, Eugen Trink 2013 - Seizure outcome in 175 patients with juvenile myoclonic epilepsy - A long-term observational study - journal Elsevier; Epilepsy Research (2014) 108, 1817–1824
Symptoms of myoclonic epilepsy
The clinical picture of myoclonic epilepsy depends on the type of disease. Among the main pathological symptoms are:
- Cramps. The disease is characterized by myoclonic seizures. They are not accompanied by severe pain. Cramps most often affect the limbs, less often the face and torso. On average, the duration of a seizure is 10-20 minutes. Consciousness is preserved.
- Loss of consciousness. Occurs extremely rarely. Characteristic of the youthful form.
- Tonic-clonic seizures. This form of seizure is accompanied by loss of consciousness and painful muscle contraction.
- Oligophrenia. Mental retardation occurs in various forms. Most often this is a disorder of creative thinking and intelligence.
- Mental disorders. Expressed by hallucinations, neuroses and borderline states.
Determining clinical symptoms is necessary to select the correct therapy. Doctors at the Yusupov Hospital develop an individual treatment plan for each patient.
Course and treatment
Children with SDMED do not experience seizures of any other type, even if they remain untreated (up to 8.5 years in one of our patients), this is especially true for petit epileptic or tonic seizures. Clinical examination results are normal. Interictal myoclonus was described only by Giovanardi-Rossi et al. (1997) in 6 patients. Analyzing the condition of our patients, we found mild interictal myoclonus in 2 according to EEG recordings. Many patients were not examined, but when CT and MRI were performed, the results were normal (33 patients).
Outcome likely depends on early diagnosis and treatment. Myoclonus is easily amenable to monotherapy with valproate, and the child’s development is appropriate for his age. If left untreated, the patient continues to have myoclonic seizures, which can lead to impaired psychomotor development and behavioral abnormalities.
Treatment methods were more or less thoroughly tested on 74 patients. 65 patients received monotherapy, 6 received polytherapy, and 3 received no treatment. Monotherapy included valproate (VPA), phenobarbital (PB), nitrazepam (NTZ). 6 patients received therapy with primidone (PRM) and ethosuximide (ESM). As a result of treatment, seizures disappeared in 69 patients (93%).
These data confirm that VPA is the first choice drug for SDMED. However, treatment should be carried out under the control of drug concentration in plasma, because improper use may lead to relapse or cause drug-resistant epilepsy.
Diagnosis of myoclonic epilepsy
Myoclonic epilepsy requires a comprehensive diagnosis. The examination includes:
- Collection of complaints and medical history. During the initial examination, the neurologist questions the patient about existing complaints, the time of their appearance, and the severity of symptoms. In addition, the duration of the attacks and the condition after them are specified.
- Neurological examination. The doctor evaluates reflexes and conducts tests to assess the state of nervous activity.
- EEG. Thanks to the study, it is possible to establish the pathological activity of various brain structures. The lesion site is determined in this way.
- MRI. To enhance the effect, the examination is carried out using contrast. Myoclonic epilepsy is not characterized by the presence of structural changes in the brain. However, there are exceptions.
The Yusupov Hospital uses modern medical equipment to diagnose myoclonic epilepsy. It allows you to quickly and effectively determine the presence of the disease. Based on the data obtained, correct therapy is prescribed.
Types of myoclonic epilepsy
Myoclonic epilepsy is divided into several types. Let's look at the main ones.
Infantile myoclonic epilepsy
Diagnosed in 30-40% of cases. Characterized by symptoms such as hyperkinesis or tics. A mild degree of development of the disease can go unnoticed for many years. Intellectual development does not suffer against the background of infantile myoclonic epilepsy. The disease can appear in a child from 2 months to 3 years.
Dravet syndrome
Identified in the first year of life. Symptomatically reminiscent of infantile myoclonic epilepsy. Dravet syndrome causes severe mental disorders. They manifest themselves as oligophrenia, mental retardation. Without correct treatment, the number of attacks increases to several times a week.
Unferricht-Lundborg disease
It is considered a genetic disease. Characterized by severe neurological symptoms. Unferricht-Lundborg disease is also accompanied by mental disorders. The first attack most often occurs during puberty. The disease is diagnosed in 10-20% of cases.
MERRF epilepsy
More often diagnosed in adults. Myoclonic epilepsy of adolescence is characterized by tonic-clonic paroxysms. The disease is not accompanied by neurological disorders or mental disorders. Consciousness remains clear during the attack.
Absence seizures are a type of epileptic seizure. Considered one of the symptoms of myoclonic epilepsy.
Depending on the progression of epilepsy, the following forms of the disease are distinguished:
- Progressive. The clinical picture of epilepsy is gradually increasing. As the disease progresses, the risk of death increases. In some cases, the disease is difficult to treat.
- Stable. Symptoms remain at approximately the same level.
- Remitting. Signs of epilepsy may develop slowly and then subside over a long period of time. Over time, complete disappearance of pathological signs is possible.
Benign myoclonic epilepsy of infancy
DMEM is diagnosed between 6 months and 3 years of age. The child's development is normal until the first myoclonic seizures appear. Approximately 20% of children present with rare seizures at birth or during the neonatal period. The patient's general condition rarely suffers; neurological disorders are not detected. The first myoclonic attacks affect the upper limbs, neck and head, and rarely the legs. They can have different intensities, including in the same child during different episodes. Severity ranges from barely noticeable twitching to visible fibrillation.
The frequency of seizures is 2-3 times a day at different time intervals. There are no long series of attacks observed. It is possible to provoke an attack with a loud sound, tactile or rhythmic light stimulation. After each episode, there may be a refractory period lasting from 20 to 120 seconds. In this time period, even intense stimulation does not cause a new attack. In this case, muscle atony is often observed. The disease is characterized by an intensification of myoclonic attacks when falling asleep (drowsiness) and their disappearance in the slow-wave sleep phase.
There are reflexive and spontaneous variants of DMEM. In the first case, myoclonic seizures develop after exposure to triggers. The spontaneous form occurs without any predictors. In the early stages of the disease and when the intensity of myoclonus is low, parents and pediatricians may mistake attacks for normal motor reactions of the child. Relatively severe myoclonic attacks can be accompanied by tilting the head forward, abductor and adduction movements, bending of the arms, and rarely by smooth rotation of the eyeballs. Often parents note a characteristic “nod” of the head lasting from 1 to 3 seconds, rarely – up to 10 seconds. (in older children). In some cases, the only clinical manifestation of DMEM is prolonged closing of the eyes.
In severe forms, generalization of convulsions is possible, accompanied by loss of balance, sudden loss of objects from hands, and rarely, disorders of consciousness. The process sometimes involves the intercostal muscles, the anterior abdominal wall and the diaphragm, which causes breathing disturbances and an expiratory noise may be heard. DMEM is characterized by an increase in the intensity of clinical manifestations up to a certain age and their subsequent complete disappearance. With a long course of the disease, a lag in psychomotor development is possible. Transformation into other forms of seizures, including absence seizures, does not occur even in the absence of specific treatment.
Treatment of myoclonic epilepsy
Medications are used to treat myoclonic epilepsy. Doctors at the Yusupov Hospital use the following groups of drugs:
- antiepileptics;
- barbiturates;
- tranquilizers;
- nootropics.
An individual treatment plan is developed for each patient. It takes into account the form of myoclonic epilepsy, stage of development, age of the patient and the presence of concomitant diseases. This approach allows you to quickly and effectively select therapy. Experienced neurologists, epileptologists, and psychiatrists conduct consultations at the Yusupov Hospital. Rehabilitation is carried out by qualified massage therapists and exercise therapy instructors. In order to make an appointment, you need to call or leave a request on the official website of the hospital.