Сахарный диабет (СД) в настоящее время является глобальной психологической, социальной и экономической проблемой. По данным IDF (2012), во всем мире СД страдают 371 млн человек. По прогнозам ВОЗ, за период 2005—2030 гг. число случаев смерти от диабета увеличится в 2 раза.
Анализ данных федерального регистра показал, что распространенность СД 1-го типа (СД1) за 10 лет у детей выросла на 35,7% (с 59,4 до 80,6% на 100 тыс. детского населения); у подростков — на 68,9% (с 108,5 до 183,5% на 100 тыс.), у взрослых — на 2,36% (с 224,5 до 229,8% на 100 тыс.) [1]. Основные тенденции в динамике эпидемиологических показателей СД1 у детей в РФ сопоставимы со средними показателями мирового среднегодового прироста заболеваемости (2,8%) [2].
Хотя СД1 составляет лишь 10% от всех больных СД, протекает он особенно тяжело и сопровождается развитием сосудистых осложнений. СД1 в детском и подростковом возрасте уже на ранних стадиях является фактором риска развития хронической цереброваскулярной патологии и проявляется как клиническими неврологическими синдромами, так и субклиническими нарушениями ЦНС, выявляющимися при нейропсихологическом, нейрофизиологическом и биохимическом исследованиях [3]. Медицинскую и социально-экономическую значимость этой проблемы трудно переоценить, учитывая распространенность СД и частоту поражений нервной системы, что приводит к колоссальным материальным затратам на лечение и социальное обеспечение таких пациентов. Частота поражений нервной системы при СД коррелирует с длительностью заболевания, его тяжестью и возрастом больных [4]. Значимая роль отводится ранней идентификации энцефалопатии (ЭП), с учетом прогрессирующего характера поражения мозговых структур. Даже легкие или умеренные нарушения двигательной, эмоционально-волевой и когнитивной сферы приводят к социальной и бытовой дезадаптации, что снижает комплаентность больных в отношении врачебных рекомендаций. Качество жизни (КЖ) больных СД и их семей является более низким, чем в общей популяции [5—7]. Параметры КЖ могут стать определяющими факторами в способности индивида управлять своим заболеванием и обеспечивать самоконтроль.
Наибольшее влияние на КЖ при СД1 в детском и подростковом возрасте оказывают психоэмоциональные особенности больных, режимы инсулинотерапии, типы отношения к болезни [8]. Негативное влияние СД1 отмечено в отношении таких аспектов КЖ, как свобода питания, трудовая деятельность, физические возможности и беспокойство о будущем. Среди факторов, опосредующих влияние СД, выделяют компенсацию углеводного обмена, осложнения, поведение пациента, связанное с заболеванием, частоту самоконтроля гликемии, а также гендерный фактор [9].
ЭП «в чистом виде» встречается лишь у больных СД1 (в 80,7% случаев), поскольку ее развитие обусловлено преимущественно неэффективным метаболическим контролем [10, 11]. Термин «диабетическая энцефалопатия» (ДЭ) предложен R. DeJong в 1950 г. Под ДЭ понимают стойкую церебральную патологию, возникающую под воздействием обменных и сосудистых нарушений, которая клинически проявляется неврозоподобными и психоподобными дефектами, органической неврологической и вегетативной симптоматикой. ДЭ включает характерные биохимические, электрофизиологические и морфологические изменения, которые могут привести к когнитивному дефициту и значительно снижают КЖ как самого больного, так и его близких [12]. По классификации Е.В. Шмидта (1985), ЭП представляет собой прогрессирующее диффузное мелкоочаговое поражение головного мозга, проявляющееся сочетанием симптомов очагового поражения и астенических симптомов. Клинический полиморфизм ДЭ дает основание предполагать существование как минимум нескольких патофизиологических механизмов ее формирования. Патогенез ДЭ связывают с двумя основными компонентами — метаболическим и сосудистым. Развитие микроангиопатии опосредовано накоплением в сосудистой стенке ЛПНП, активизацией процессов ПОЛ, увеличением образования свободных радикалов, подавлением синтеза простациклина, обладающего антиагрегантным и сосудорасширяющим действием [12, 13]. Прогрессирование микроангиопатии приводит к снижению эндоневрального кровотока с развитием гипоксии, способствующей переключению энергетического метаболизма нервной ткани на малоэффективный анаэробный гликолиз, в процессе которого из одной молекулы глюкозы образуется лишь две молекулы АТФ, тогда как в реакции аэробного гликолиза — 38 молекул. В результате в нейронах снижается концентрация фосфокреатинина, возрастает содержание лактата, что приводит к развитию кислородного и энергетического голодания нервной ткани. Снижение эндоневральной микроциркуляции и усугубление нарушений функции нервных волокон способствует уменьшению синтеза и увеличению разрушения оксида азота (NO), обладающего вазодилатирующим действием, что может стать одной из причин развития артериального спазма, являющегося важным патогенетическим механизмом развития артериальной гипертензии при СД. Помимо патогенетической значимости нарушений эндоневрального кровотока, важную роль играют и метаболические расстройства. Установлено, что снижение скорости проведения импульса по миелиновым волокнам обусловлено патологически высокой внутриаксональной концентрацией ионов Na+, в развитии которой основная роль принадлежит снижению активности тканевой Na+/К+-АТФазы, что вызывает вторичные сосудистые нарушения, расстройства нейротрофики, нейротоксикоз и, как следствие, структурное изменение нейронов, а также нарушение скорости проведения возбуждения по нерву [12]. СД вносит большой вклад в потерю белого вещества головного мозга [13, 14].
Не вызывает сомнений взаимосвязь остро или хронически развивающихся изменений ЦНС с гипер- или гипогликемией. М.Р. Чуйко и соавт. [16] показали, что клинические проявления ЭП особенно характерны для пациентов с частыми гипогликемическими состояниями, дебютируя в возрасте 26—35 лет. I. Brands [10] в своей книге «Diabetes and the brain» описал одну из патогенетических причин когнитивных расстройств при СД, которой является гипергликемия.
На моделях животных с индуцированным СД1 показано, что дефицит инсулина играет важную роль в апоптозе нейронов, а также в развитии дегенеративных явлений в белом веществе головного мозга [17]. Базисными механизмами, лежащими в основе осложнений СД, являются активизация полиолового и гексозаминового путей метаболизма глюкозы, образование конечных продуктов избыточного гликирования белков, повышение содержания различных изоформ протеинкиназы С. Большинство известных метаболических и сосудистых механизмов развития экстра- и интрацеллюлярной патологии при поздних осложнениях СД обеспечивают гиперпродукцию супероксида в митохондриях. Таким образом, основной причиной формирования поздних осложнений, в том числе и ЭП, является оксидантный стресс — нарушение равновесия между продукцией свободных радикалов и активностью антиоксидантных ферментов, которое при СД снижено.
Для диагностики ЭП чаще используются психологические опросники, которые не исключают субъективизм в оценке результатов. Поэтому ведется поиск диагностических биомаркеров повреждения головного мозга [10, 17, 31, 32]. В настоящее временя такими биомаркерами считают нейроспецифические белки (НСБ), которые изменяются количественно при различных заболеваниях [18]. Среди них наиболее изученными являются: нейроспецифическая енолаза (НСЕ) — маркер нейронов, глиальный фибриллярный кислый белок (GFAP), белок S100 (изоформы — S100A1 и S100B) — маркеры гибели астроцитов и основной белок миелина (МВР) — маркер повреждения олигодендроцитов. Повышение НСБ в крови указывает на повреждение нервной ткани и позволяет дать прижизненную характеристику состояния ЦНС и оценить динамику нейродегенеративного процесса [19].
В работах B. Danna и соавт. [20] имеются сведения о роли белков S100A1 и S100B в развитии ЭП при СД. В последние годы определение этих белков все более активно используется для диагностики повреждения мозговой ткани при нарушениях мозгового кровообращения. Повышение концентрации S100A1 и S100B коррелирует с объемом поражения мозга. Белок S100B продуцируется преимущественно астроцитами мозга и является маркером активации астроглии, опосредующим свои эффекты взаимодействием с рецепторами конечных продуктов гликозилирования. Показано, что S100B проявляет нейротрофическую активность при физиологической концентрации и нейротоксическую — при высокой концентрации. Белок S100B рассматривается как один из узловых молекулярных компонентов сложных внутриклеточных систем, обеспечивающих функциональный гомеостаз клеток мозга путем сопряжения и интеграции разноплановых метаболических процессов [21].
Таким образом, изоформы белка S100 представляют собой наиболее универсальные из известных макромолекул, участвующих в регуляции практически всех основных мембранных, цитоплазматических и ядерных метаболических процессов, связанных с обеспечением механизмов восприятия и интеграции поступающей в нервную систему информации [21, 22], в ответе генов раннего реагирования, в реализации генетических программ апоптоза и антиапоптозной защиты [23]. У трансгенных мышей с гиперпродукцией S100B выявлены дефекты функции гиппокампа и нарушение кратковременной памяти, частичное снижение способности решать пространственные задачи [24—27]; кроме того, у них нарушена адаптация к новой обстановке, но при этом усиливается редукция тревоги (по данным ряда поведенческих тестов) [27—29, 31]. M. Rothermundt и соавт. [29] показали, что уровень S100B повышен у больных с легкой/умеренной депрессией, его содержание в сыворотке увеличивалось и у пациентов с меланхолическим подтипом депрессии в отличие от немеланхолического [31]. Повышение уровня S100B в крови у больных СД можно рассматривать как результат развития реактивного глиоза и повышенной проницаемости гематоэнцефалического барьера, вызванных гипергликемией и церебральной гипоксией. Таким образом, повышение уровня S100В связано преимущественно с дисциркуляторными нарушениями, что с высокой степенью вероятности подтверждает преимущественную активацию реактивного глиоза при СД. Следовательно, тяжесть поражения ЦНС при данном заболевании в определенной мере обусловлена особенностями реакции структур мозга на гипоксию и метаболический стресс, делая определенные участки ЦНС (прежде всего мезэнцефальные) наиболее уязвимыми при прочих равных условиях, особенно при кетоацидозе, что подтверждают Е. McIntyre и соавт. [30], которые рекомендуют использовать S100 в качестве биомаркера повреждения головного мозга.
О.З. Пузикова и соавт. [32] нашли, что высокое содержание белка S100В в сыворотке детей и подростков с СД находится в прямой зависимости от степени выраженности церебральных нарушений и уровня десатурации в период ночного сна, а также от времени проведения слухового импульса в срединных структурах мозга. Между уровнем этого белка и скоростью кровотока в передней и задней мозговых артериях обнаружена обратная зависимость, что свидетельствует о связи усиленного реактивного астроцитоза при СД с гипоксическими, дисциркуляторными процессами и ухудшением нейрофизиологических параметров в ЦНС. M. Strachan и соавт. [33] показали, что концентрация НСЕ и S100 может играть прогностическую роль в развитии неврологического дефицита. Так, у 2 из 3 пациентов, которые умерли вследствие эпизодов гипогликемии, концентрации этих маркеров были значительно повышены. M. Hovsepyan и соавт. [34] нашли, что сывороточные уровни S100B и НСЕ при СД1 не имели существенных различий, однако было отмечено значительное увеличение антител к НСЕ.
Ключевым патогенетическим механизмом ЭП некоторые авторы считают развитие дисрегуляции серотонинергической, норадренергической систем, а также системы эндогенных опиоидных пептидов мозга. Томскими учеными [35] получены результаты, свидетельствующие о повышении уровня НСЕ в 1,64 раза и уменьшении серотонина в 1,66 раза в крови у пациентов с СД1 по сравнению с контролем. Выраженность нарушений зависела от степени компенсации углеводного обмена. Авторы [35] предположили, что хроническая гипергликемия при СД1 способствует формированию условий для повреждения не только клеток эндотелия сосудов, но и цитомембран клеток мозга, что сопровождается появлением в крови некоторых специфических для мозга пептидов, в частности НСЕ. Повышенный уровень НСЕ в крови являлся доказательством повреждения нейрональных мембран, свидетельствуя о дисфункции мозговых структур, усугубляющейся с увеличением длительности СД1.
К.А. Павлов [36] опубликовал сведения о белке промежуточных филаментов астроцитов (GFAP), который высоко специфичен для мозга и высвобождается только в случае некротической смерти клеток и цитолиза. Практически любой патологический процесс в ЦНС приводит к выраженной активации астроглиального компонента нервной ткани. Наиболее ярким проявлением реактивного астроглиоза на молекулярно-биологическом уровне является резкое увеличение экспрессии GFAP в активированных астроцитах. Дальнейшее развитие патологического процесса приводит к гибели реактивных астроцитов, вследствие чего нарушается резистентность клеточной мембраны, и GFAP оказывается в межклеточной жидкости, откуда элиминируется в кровоток и ликвор пациента. Появление GFAP в биологических жидкостях организма возможно лишь при нарушении резистентности гематоэнцефалического барьера и несостоятельности эндотелия сосудов [37, 38]. Уровень GFAP в биологических жидкостях напрямую зависит от количества погибших или поврежденных астроцитов, что, в свою очередь, отражает степень выраженности нейродегенераторного процесса [39]. E. Coleman и соавт. [40] показали, что при стрептозоциновом диабете у крыс возрастает уровень GFAP в гипокампе, мозжечке и белом веществе. С другой стороны, индуцированный СД in vivo ингибировал активность астроцитов и, тем самым, уменьшал уровень GFAP [41]. Колебания уровня данного белка зависят от длительности патологического процесса [42]. Данные о содержании GFAP в различных областях головного мозга в эксперименте неоднозначны [43, 44]. Таким образом, роль GFAP в качестве маркера остается недостаточно изученной.
В настоящее время активно изучается такой маркер повреждения головного мозга как MBP.
В.П. Чехонин и соавт. [45] нашли высокий уровень МВР у детей с ЭП. В исследовании итальянских ученых впервые сообщается, что содержание двух изоформ МВP (18,5 и 21,5 кДа) уменьшается в спинном мозге у крыс со стрептозоциновым диабетом [46]. При СД1 наблюдалось повышение уровня аутоантител к МВР класса IgG и IgM, которое зависело от длительности заболевания [47].
Таким образом, несмотря на разнообразие маркеров повреждения головного мозга, их роль в развитии и прогрессировании ЭП при СД не установлена. Необходимы дальнейшие исследования с целью создания алгоритма диагностики этого состояния [48—50].
Диабетическая энцефалопатия
Поражение нервной системы является одним из наиболее ранних и частых осложнений сахарного диабета. Диабетическая энцефалопатия относится к центральной форме нейропатии при сахарном диабете.
По данным разных авторов, эта патология встречается у 90-100% больных сахарным диабетом (СД). Более того, в ряде случаев симптомы со стороны нервной системы предшествуют появлению клинических признаков СД.
После открытия сахарного диабета как самостоятельного заболевания в XVIII веке и появления первых работ по нейродиабетологии интерес к изучению патологии нервной системы неуклонно растет.
Медицинскую и социально-экономическую значимость этой проблемы трудно переоценить, учитывая распространенность сахарного диабета, частоту вызываемого им развития инвалидизирующих поражений нервной системы со снижением качества жизни и социальной активности больных, а также колоссальные материальные затраты на лечение и социальное обеспечение пациентов. Частота поражений нервной системы при сахарном диабете коррелирует с длительностью заболевания, степенью тяжести и возрастом больных.
При сахарном диабете поражаются центральная нервная система (энцефалопатия, миелопатия), периферическая нервная система (поли- и мононейропатии), периферическая вегетативная нервная система (автономная нейропатия).
Термин «диабетическая энцефалопатия» (ДЭ) предложен R. DeJong в 1950 году. Под диабетической энцефалопатией понимали стойкую церебральную патологию, возникающую под воздействием острых, подострых и хронических диабетических обменных и сосудистых нарушений, клинически проявляющихся неврозоподобными и психоподобными дефектами, органической неврологической и вегетативной церебральной симптоматикой.
По классификации Е. В. Шмидта (1985) энцефалопатия представляет собой прогрессирующее диффузное мелкоочаговое поражение головного мозга, клинически выраженное сочетанием симптомов очагового поражения головного мозга и астенических проявлений.
Этиология и патогенез диабетической энцефалопатии
Диабетическая энцефалопатия, как и другие виды диабетической нейропатии, развивается вследствие распространенного поражения нейронов и их отростков. Чрезвычайный клинический полиморфизм диабетической энцефалопатии дает основание предполагать существование как минимум нескольких патобиохимических механизмов, участвующих в ее формировании, между которыми, очевидно, существуют взаимосвязи. Исследования последних лет подтверждают эти предположения, однако детальное изучение продолжается.
Патогенез диабетической энцефалопатии традиционно связывается с двумя основными направлениями – метаболическим и сосудистым. При этом несомненный приоритет признается за нарушениями микроциркуляции – диабетической микроангиопатией.
Развитие микроангиопатии связано с накоплением в сосудистой стенке липопротеинов низкой плотности (ЛПНП), активизацией процессов перекисного окисления липидов (ПОЛ), увеличением образования свободных радикалов, подавлением синтеза простациклина, обладающего антиагрегантным и сосудорасширяющим действием. Прогрессирование микроангиопатии приводит к снижению эндоневрального кровотока.
Развивающаяся гипоксия переключает энергетический метаболизм нервной ткани на малоэффективный анаэробный гликолиз, в процессе которого из одной молекулы глюкозы образуется лишь две молекулы АТФ, в то время как в реакции аэробного гликолиза – 38 молекул. В результате в нейронах снижается концентрация фосфокреатина, возрастает содержание лактата, развивается кислородное и энергетическое голодание нервной ткани.
Снижению эндоневральной микроциркуляции и усугублению нарушений функции нервов способствуют уменьшение синтеза и увеличение разрушения NO, обладающего вазодилатирующим действием. Это может стать одной из причин развития артериального спазма, являющегося важным патогенетическим механизмом развития артериальной гипертензии у больных сахарным диабетом.
Признавая патогенетическую значимость нарушений эндоневрального кровотока в формировании диабетической энцефалопатии, нельзя недооценивать и важность метаболических расстройств. Установлено, что снижение скорости проведения по миелиновым волокнам обусловлено патологически высокой внутриаксональной концентрацией ионов Na+, в развитии которой основная роль принадлежит снижению активности тканевой Na+/К+-АТФазы.
Предопределяющими факторами развития диабетической энцефалопатии являются:
- патологические биохимические процессы, запускаемые в условиях абсолютного или относительного дефицита инсулина: активизация полиолового пути окисления глюкозы, истощение запасов миоинозитола (предшественник фосфоинозитола –основного регулятора активности тканевой Na+-К+-АТФазы);
- повышение в нейронах концентрации свободных радикалов, лимитирующих процессы ПОЛ и окислительный стресс – важнейший фактор повреждения нервных клеток; необратимое гликозилирование белков – включение углеводов в структуру белков сыворотки крови, клеточных мембран, липопротеидов, коллагена, нейронов, приводящее к нарушению функциональной активности клеток, образованию аутоантител к белкам сосудистых стенок (существенный фактор патогенеза микроангиопатии);
- образование устойчивого комплекса HbA1c, обладающего низким сродством к кислороду, вследствие чего возникает тканевая гипоксия.
Все это вызывает вторичные сосудистые нарушения, расстройства нейротрофики, нейротоксикоз и, как следствие, структурное изменение нейронов и нарушение скорости проведения возбуждения по нерву.
Гипергликемия − несомненно важный фактор, способствующий развитию диабетической энцефалопатии посредством многообразных обменных нарушений, которые она индуцирует. Однако убедительных доказательств непосредственной связи между гипергликемией и диабетической энцефалопатии до сих пор не получено. На современном этапе развития нейродиабетологии практически не осталось сомнений в том, что достижение стабильной нормогликемии не позволяет достичь прекращения прогрессирования патологии нервной системы. Предполагают, что для развития диабетической энцефалопатии необходимым условием являются метаболические нарушения, а основой для их проявления служит генетическая предрасположенность.
Также в патогенезе поражения центральной нервной системы при сахарном диабете и развитии диабетической энцефалопатии большое значение имеют дислипидемия и атеросклероз, макроангиопатия и артериальныя гипертензия, механизмы патогенеза которых тесно взаимосвязаны.
Клиническая картина и диагностика диабетической энцефалопатии
Диабетическая энцефалопатия обычно развивается постепенно, часто течет субклинически, ее проявления маскируются: у молодых людей – последствиями перенесенных острых кетоацидотических эпизодов, у пожилых – нарушениями мозгового кровообращения.
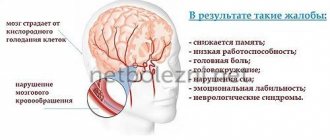
В клинической картине, прежде всего, наблюдается астенический синдром (общая слабость, повышенная утомляемость, снижение работоспособности, эмоциональная лабильность, тревожность, нарушение концентрации внимания, бессонница).
Часто выявляется цефалгический синдром, проявляющийся головной болью. При этом больные чаще всего описывают ее как сжимающая, стискивающая, по типу «тесного головного убора (головная боль напряжения), или как чувство тяжелой головы и невозможности сосредоточиться (ишемически-гипоксическая головная боль).
У пациентов практически всегда отмечается синдром вегетативнойдистонии с развитием вегетативных пароксизмов, предобморочных состояний и обмороков. Кроме астенических и вегетативно-дистонических проявлений обнаруживаются очаговые нарушения: верхнестволовые (анизокория, расстройство конвергенции, признаки пирамидной недостаточности), вестибулярно-атаксический синдром (головокружение, шаткость походки, отклонения при выполнении проб на координацию движений).
Довольно характерным в клинической картине диабетической энцефалопатии является наличие расстройств когнитивных функций: нарушение памяти, внимания, замедление мышления, апатия, депрессия, указывающие на преимущественную дисфункцию неспецифических срединных структур головного мозга. При сахарном диабете часто встречается депрессия: как показывают наблюдения, более 32% пациентов, страдающих сахарным диабетом, подвержены депрессии. Кроме влияния на общее самочувствие, депрессия опасна потерей контроля над течением самого заболевания, правильным питанием и применением инсулина. По мнению специалистов, одной из причин склонности больных сахарным диабетом к депрессии являются некоторые биохимические изменения в организме. Другая причина – постоянная зависимость людей, обусловленная болезнью.
У части больных сахарным диабетом в результате гипогликемических состояний развивается гипогликемическая энцефалопатия. Клинически она проявляется нарастанием вялости, апатии, адинамии после физической работы и натощак, расстройством сознания, чаще всего по типу делирия. Характерно наличие судорожного синдрома, возможны пирамидные гемипарезы.
Для постановки диагноза энцефалопатии, кроме астенических и вегетативно-дистонических жалоб, необходимо установление у больного очаговой неврологической симптоматики. Изменения электроэнцефалограммы (ЭЭГ) у пациентов с диабетической энцефалопатии характеризуются как дисрегуляторные, носят диффузный характер (в виде различных «уплощений» ЭЭГ, гиперсинхронизационных ритмов, редукций альфа-ритма локальных и общих, вкраплений патологических волн тета- и дельта-типов, изменений реактивности ЭЭГ кривых и т.д.).
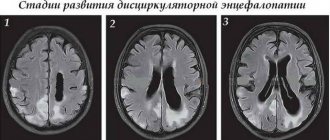
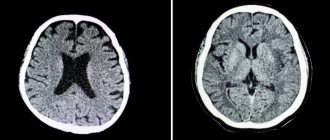
У больных сахарным диабетом пожилого возраста диабетическая энцефалопатия с большой частотой сопровождается очаговым неврологическим дефицитом, макроструктурными изменениями головного мозга (атрофия, постинсультные изменения), объективизируемыми при компьютерной томографии (КТ) и магнитно-резонансной томографии (МРТ), которые являются проявлением характерных для сахарного диабета и патогенетически связанных с ним макроангиопатий, атеросклероза и АГ.
Инсульт и транзиторные ишемические атаки с этих позиций могут рассматриваться как проявления центральной нейропатии.
Лечение диабетической энцефалопатии
Лечение диабетической энцефалопатии включает адекватный контроль гликемии, вазоактивную и метаболическую терапию.
Достижение стойкой компенсации СД является необходимым, но недостаточным условием предупреждения и лечения диабетической энцефалопатии. Для больных сахарным диабетом 2-го типа (СД 2) это особенно существенно, поскольку при нем тканевые обменные нарушения детерминированы генетически и сохраняются постоянно, даже в условиях контролируемой нормогликемии, а следовательно, проведение перманентной профилактической терапии, направленной на улучшение метаболизма нервной ткани, патогенетически обосновано.
Метаболическая терапия. С целью нормализации нейрометаболических процессов при сахарном диабете в последние годы в нашей стране и за рубежом наиболее широко используются антиоксиданты. Приоритетом в этой группе средств пользуются препараты α-липоевой (тиоктовой) кислоты (тиоктацид, тиогамма, диалипон, эспалипон, берлитион, тиоктодар), обладающие мощным антиоксидантным действием и занимающие одно из центральных мест в метаболической терапии. Лечение тиоктовой кислотой в результате ее позитивного влияния на основные звенья патогенеза приводит к улучшению энергетического метаболизма нервной ткани, увеличению продукции АТФ и трансмембранного транспорта ионов вследствие активации митохондриальных окислительных процессов, что определяет перспективность ее использования.
В лечении диабетической энцефалопатии на сегодняшний день активно используется моделирование новых комбинированных препаратов, в том числе и церебропротекторного действия, в частности ― пирацетама с тиотриазолином. Фармакологический эффект тиоцетама обуславливается взаимопотенциирующим действием тиотриазолина и пирацетама.
Тиотриазолин обладает соответствующими фармакологическими (высокая антиоксидантная, противоишемическая активность), фармакологическими (совместимость с другими препаратами) и фармакоэкономическими характеристиками.
Под нашим наблюдением находились 50 пациентов (36 женщин и 14 мужчин) в возрасте от 34 до 65 лет как с СД 1, так и СД 2. Из них 21 человек – с СД 1 и 29 – с СД 2 с сопутствующей энцефалопатией как смешанного, так и метаболического генеза. Длительность сахарного диабета от 1 года до 20 лет: 10 человек болели сахарным диабетом – 1-2 года, 12 человек – от 3 до 5 лет и 28 человек – от 10 до 20 лет. Все пациенты получали стандартную комплексную терапию, включающую также и тиоцетам, который вводили по 5,0 мл в/в в течение 10 дней, затем в течение месяца пациенты принимали тиоцетам по 1 табл. трижды в день за 30 мин. до еды.
В процессе наблюдения у каждого пациента оценивали соматический и неврологический статус, проводился мониторинг лабораторных показателей с целью оценки общего влияния препарата на организм пациента (в начале и в конце исследования). Клиническая оценка тиоцетама оценивалась путем двукратного (перед началом лечения и на 38-й день) проведения реоэнцефалограммы (РЭГ) с использованием компьютерного диагностического комплекса «Реокомп», также проводили исследование когнитивных и мнестических функций.
Данные динамического исследования показателей когнитивных функций выявили следующие особенности. Так, отмечено статистически значимое положительное влияние тиоцетама на все исследуемые функции: увеличение показателя «количество строк» на 27% (Р < 0,05), уменьшение доли ошибок на 32% (Р < 0,05), ускорение выполнения пробы Шульте на 32% (Р < 0,01).
Анализ показателей свидетельствует о позитивном влиянии препарата тиоцетам на основные виды памяти, причем статистически значимым является влияние на моторную и оперативную память, в то время как изменения зрительной и вербальной памяти, будучи позитивными в количественном отношении, не имели статистической значимости.
Количественные характеристики позитивных статистически значимых влияний тиоцетама следующие: улучшение показателей моторной памяти на 43% (Р < 0,01), оперативной, эмоциональной, опосредованной – на 41% (Р < 0,05), для группы 2 – соответственно 16 и 18% (Р < 0,05).
В комплексной терапии диабетической энцефалопатии широко используют витамины А, С, Е, обладающие антигипоксантным действием. Нейротропной активностью обладают витамины В1, В6, В12, повторные курсы лечения которыми желательно проводить не реже двух раз в год. В последние годы появились новые, высокоэффективные формы этих средств, в частности – биологически активная жирорастворимая форма тиамина (витамин В1) – бенфотиамин, активность которого по сравнению с водорастворимыми формами тиамина выше в 8-10 раз, а возможности проникновения в нервную клетку и конвертирования в активный метаболит тиамина еще выше.
Широкое распространение в нейродиабетологии получил комбинированный препарат, включающий все витамины группы В в необходимой лечебной дозировке – нейромультивит. Одним из современных высокоактивных препаратов является Мильгамма 100 (100 мг бенфотиамина + 100 мг пиридоксина гидрохлорида) для перорального приема.
Вазоактивная терапия. Одно из главных мест в терапии микро- и макроангиопатий занимает пентоксифиллин (трентал), действие которого заключается в нормализации кровотока на капиллярном уровне за счет снижения агрегации форменных элементов крови, понижения ее вязкости и повышения способности эритроцитов к деформации. Он улучшает микроциркуляцию, способствует выведению продуктов метаболизма и токсинов, увеличению объема циркулирующей жидкости и нормализации диуреза. Пролонгированная форма пентоксифиллина позволяет создать длительное и более равномерное насыщение препаратом.
Для лечения и профилактики расстройств церебрального кровообращения используются вазоактивные препараты разных групп с преимущественно центральным действием: кавинтон, стугерон (циннаризин), сермион (ницерголин), нимодипин (нимотоп), а также инстенон – препарат с комбинированным ноотропным и церебральным вазодилатирущим действием и другие. Существенное значение в предупреждении прогрессирования ДЭ придается лечению АГ и атеросклероза.
Из монографии «Сахарный диабет: от ребенка до взрослого»
Сенаторова А.С., Караченцев Ю.И., Кравчун Н.А., Казаков А.В., Рига Е.А., Макеева Н.И., Чайченко Т.В. ГУ «Институт проблем эндокринной патологии им. В.Я. Данилевского АМН Украины» Харьковский национальный медицинский университет Харьковская медицинская академия последипломного образования МЗ Украины