Об эпилепсии мы обычно слышим не так часто, как, скажем, об инфарктах и инсультах. Однако практически каждый из нас, так или иначе, знаком с этим заболеванием если не лично, то по рассказам окружающих, у которых данным недугом страдают родственники или знакомые. И это неслучайно, ведь эпилепсию наблюдают почти у 5% населения нашей планеты, причем у детей это заболевание регистрируется втрое чаще по сравнению с взрослыми. А общий термин «эпилепсия», по заключению современных практикующих неврологов, объединяет в себе более полусотни отличных по клиническим проявлениям и лечению заболеваний, включая и наиболее распространенную генерализованную эпилепсию.
Что такое генерализованная эпилепсия
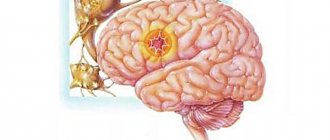
Генерализованная эпилепсия – это хроническая патология головного мозга, которая характеризуется формированием (в обоих полушариях) нейронных импульсов чрезмерной интенсивности. Во время возникающих вследствие этого приступов (которые становятся повторяющимися) временно нарушаются многие органические функции больного:
- двигательные
- вегетативные
- чувствительные
- психические
Под генерализованной эпилепсией сегодня подразумевают все эпилептические формы, основанные на стереотипности возникновения эпиприступов. Последние состоят из абсансов (кратковременного, буквально на несколько секунд, выключения сознания), генерализованных миоклонических (связанных с непроизвольным мышечным напряжением) и тонико-клонических (связанных со сменой мышечного напряжения и прерывистых мышечных сокращений) пароксизмов.
Юношеская миоклоническая эпилепсия.
Юношеская миоклоническая эпилепсия (ЮМЭ) – одна из форм идиопатической генерализованной эпилепсии, характеризующаяся дебютом в подростковом возрасте с возникновением массивных билатеральных миоклонических приступов, преимущественно в руках, в период после пробуждения пациентов. ЮМЭ – одна из первых форм эпилепсии с известным генетическим дефектом. Предполагается двулокусная модель наследования (доминантно-рецессивная), причем доминантный ген локализован на коротком плече хромосомы 6. Дебют ЮМЭ варьирует от 7 до 21 года с максимумом в возрастном интервале 11-15 лет. Заболевание может начаться в более раннем возрасте с абсансов или ГСП, с последующим присоединением миоклонических приступов в пубертатном периоде. Миоклонические приступы характеризуются молниеносными подергиваниями различных групп мышц; они чаще двухсторонние, симметричные, единичные или множественные, меняющиеся по амплитуде. Локализуются, главным образом, в плечевом поясе и руках, преимущественно в разгибательных группах мышц. Во время приступов больные роняют предметы из рук или отбрасывают их далеко в сторону. У 40 % пациентов миоклонические приступы захватывают и мышцы ног, при этом больной ощущает как бы внезапный удар под колени и слегка приседает или падает (миоклонически-астатические приступы); затем тут же встает. Сознание во время приступов обычно сохранено. Миоклонические приступы возникают или учащаются в утренние часы, после пробуждения пациента. В 90 % случаев они сочетаются с ГСП пробуждения и в 40 % – с абсансами. Основными провоцирующими приступы факторами являются депривация сна и внезапное насильственное пробуждение. Примерно 1/3 больных ЮМЭ (чаще женского пола) обнаруживают фотосенситивность. Эпилептическая активность на ЭЭГ выявляется у 85% больных в межприступном периоде. Наиболее типична генерализованная быстрая (от 4-х Гц и выше) полипик-волновая активность в виде коротких вспышек. Возможно также появление пик-волновой активности 3 Гц. Лечение. Наряду с медикаментозной терапией необходимо строго придерживаться соблюдения режима сна и бодрствования; избегать недосыпания и факторов фотостимуляции в быту. Базовые препараты – исключительно производные вальпроевой кислоты. Средняя суточная дозировка – 40-60 мг/кг. При недостаточной эффективности назначается политерапия: депакин + суксилеп (при резистентных абсансах); депакин + фенобарбитал или гексамидин (при резистентных ГСП); депакин + ламиктал или клоназепам (при резистентных миоклонических приступах и выраженной фотосенситивности). Полная медикаментозная ремиссия достигается у 75% больных, причем в большинстве случаев на монотерапии вальпроатами. Однако в последующем при отмене АЭП, рецидивы констатируются у половины больных. При данной форме эпилепсии рекомендуется отмена АЭП спустя не менее 4-х лет с момента наступления ремиссии.
Какие виды генерализованной эпилепсии известны
В зависимости от характера происхождения, генерализованную эпилепсию обычно разделяют на:
- идиопатическую – передающуюся генетически и, в основном, возникающую в детском, либо подростковом возрасте (или с возможным проявлением уже во взрослом возрасте), характеризующуюся отсутствием патологий нервной системы и, в частности, структурных изменений головного мозга (кроме изменений, регистрируемых при помощи ЭЭГ и МРТ);
- симптоматическую – возникающую вследствие черепно-мозговых травм (в большинстве случаев – при родах), либо в результате обширных органических поражений головного мозга после нейроинтоксикаций, инфекционных заболеваний, а также на фоне наследственных патологий, новообразований, некоторых дисметаболических состояний (включая внутриутробную гипоксию плода);
- криптогенную – с неустановленной этиологией.
В зависимости от присутствия в приступах судорог, генерализованная эпилепсия разделяется на:
- бессудорожную (характеризуется наличием в приступах абсансов);
- судорожную (характеризуется тонико-клоническими фазами приступа).
Идиопатическая генерализованная эпилепсия
Для всех форм идиопатической эпилепсии характерно:
- Генетическая предрасположенность (часто наличие семейных случаев эпилепсии).
- Лимитированный возраст дебюта заболевания.
- Отсутствие изменений в неврологическом статусе.
- Нормальный интеллект пациентов.
- Сохранность основного ритма на ЭЭГ.
- Отсутствие структурных изменений в мозге при нейровизуализации.
- Препараты выбора в лечении – производные вальпроевой кислоты.
- Относительно благоприятный прогноз с достижением терапевтической ремиссии в подавляющем большинстве случаев.
Каковы симптомы генерализованной эпилепсии
Симптоматика генерализованной эпилепсии обычно зависит от ее формы.
ИдиопатическаяСимптоматическая
Возраст первого проявления Детский и юношеский период (преимущественно до 21 года).Любой возрастной период, но для врожденных и наследственных патологий характерны проявления в раннем детстве.
Неврологический статус Симптоматика рассеянная, изредка – очаговая.Зависит от основного заболевания, которое определяет общемозговую и очаговую симптоматику.
Характер приступов Эпилептические пароксизмы первично-генерализованного характера: с миоклоническими судорогами (кратковременными подергиваниями конечностей), абсансами (с отсутствием внимания), генерализованными тонико-клоническими судорогами (мышечным напряжением, сменяющимся на фоне отсутствия сознания прерывистыми мышечными сокращениями, что нередко заканчивается непроизвольным мочеиспусканием и мышечным расслаблением).Генерализованные эпиприступы (на фоне клинических проявлений основного заболевания). Характеризуются общим мышечным напряжением с кратковременной остановкой дыхания, сменяющимся судорогами, которые могут продолжаться от 10 сек до 20 мин.
Когнитивные проявления Умственные способности в норме, хотя иногда могут временно немного снижаться (примерно у 5-8% пациентов).Наблюдается снижение интеллектуальных способностей, у детей может развиться олигофрения, церебральный паралич.
Юношеская абсанс эпилепсия.
Юношеская абсанс эпилепсия (ЮАЭ) – разновидность идиопатической генерализованной эпилепсии, которая характеризуется основным видом приступов – абсансами, дебютирующими в пубертатном периоде с высокой вероятностью присоединения ГСП и характерными ЭЭГ-изменениями в виде генерализованной пик-волновой активности с частотой 3 Гц и более. Дебют абсансов при ЮАЭ варьирует от 9 до 21 года, в среднем, 12.5 лет. У значительного большинства больных (75%) абсансы начинаются в сравнительно коротком временном промежутке – 9-13 лет. Важной особенностью ЮАЭ является частый дебют заболевания с ГСП – 40% случаев. Абсансы у пациентов, страдающих ЮАЭ, проявляются коротким выключением сознания с застыванием и гипомимией. Характерно значительное преобладание простых абсансов, то есть приступов без какого — либо двигательного компонента. Продолжительность приступов составляет от 2 до 30 сек, в среднем, 5-7 сек. Вместе с тем, у половины пациентов отмечаются очень короткие абсансы, не превышающие 3 сек. Характерной особенностью ЮАЭ является и относительно невысокая частота приступов по сравнению с ДАЭ. У большинства больных преобладают единичные абсансы в течение дня или 1 приступ в 2-3 дня. Генерализованные судорожные приступы констатируются у большинства больных – 75%. В группе больных с ГСП заболевание чаще дебютирует не с абсансов, а с тонико-клонических судорожных пароксизмов. ГСП характеризуются короткими нечастыми тонико-клоническими судорогами, возникающими, обычно, при пробуждении или засыпании. В отличие от ДАЭ, гипервентиляция провоцирует возникновения абсансов не более чем у 10% больных ЮАЭ. ГСП у 20% больных провоцируются депривацией сна. При ЭЭГ-исследовании в межприступном периоде результаты, близкие к норме, констатируются у 25% пациентов. Основным ЭЭГ-паттерном является генерализованная пик-волновая активность с частотой 3 Гц и более (4-5 в сек), носящая, преимущественно, симметричный и билатерально- синхронный характер. Лечение. Эффективность лечения при ЮАЭ достоверно ниже, чем при ДАЭ. Терапевтическая ремиссия достигается, в среднем, у 60% больных, значительное урежение приступов – 35%, отсутствие эффекта – 5%. Лечение начинается с монотерапии препаратами вальпроевой кислоты. Средняя дозировка составляет 30-50 мг/кг/сут. Ввиду крайне высокой вероятности при ЮАЭ присоединения ГСП, начинать лечение с сукцинимидов, а также применять их в виде монотерапии, категорически противопоказано. При отсутствии существенного эффекта от монотерапии вальпроатами в достаточно высоких дозах, применяется комбинация вальпроатов с сукцинимидами или ламикталом. Средние дозы суксилепа составляют 20 мг/кг/сут.; ламиктала – 1-5 мг/кг/сут.
В чем заключается диагностика генерализованной эпилепсии
Генерализованная эпилепсия диагностируется доктором на основании:
- тщательного сбора анамнеза (включая информацию о наличии заболевания в роду, возрасте больного, в котором наблюдался первый приступ, длительности и характере приступа, наличии судорог)
- лабораторного исследования крови и мочи
- эхоэнцефалографии (включая ЭЭГ мониторинг)
Также дополнительно для уточнения диагноза могут понадобиться:
- КТ
- ЯМРТ
- биохимическое исследование крови и ликвора
- консультации других специалистов (нейрохирурга, инфекциониста, детского офтальмолога)
Каково лечение генерализованной эпилепсии
Эффективное лечение данного вида эпилепсии напрямую зависит от формы недуга, а также характера приступов. И если оно не всегда позволяет забыть пациенту о его недуге, в идеале оно должно быть направлено на максимальное снижение частоты и силы приступов.
Антиконвульсантную терапию генерализованной эпилепсии обычно рекомендовано начинать не ранее прохождения второго приступа и только при условии установления точного диагноза. Начинают ее обычно назначением базового препарата именно для этой эпиформы (например, вальпроатов и др.), стартуя с дозы, составляющей примерно четверть от стандартной терапевтической. В течение последующих двух-трех недель (при непременном условии хорошей переносимости и отсутствии лечебного эффекта) стартовую дозу увеличивают еще наполовину, доводя далее до средней терапевтической. Такая монотерапия обычно достаточно эффективна в случае идиопатической генерализованной эпилепсии.
Если в результате лечения положительная динамика не наблюдается, либо состояние ухудшают различные побочные эффекты, препарат может быть заменен доктором на другой. Успешной терапия может считаться при наступлении полной ремиссии (без приступов) в течение трех лет с начала лечения, после чего возможно снижение дозировки.
Генерализованная эпилепсия сегодня – это не приговор пациенту, ведь вовремя поставленный правильный диагноз и эффективная терапия позволяет больному значительно улучшить качество своей жизни уже на первых этапах лечения. Вас или Ваших близких беспокоит этот недуг? Опытные специалисты медицинского всегда готовы оказать Вам необходимую помощь и дать возможность с уверенностью смотреть в завтрашний день!
Эпилепсия с изолированными генерализованными судорожными приступами.
Эпилепсия с изолированными ГСП определяется как синдром идиопатической генерализованной эпилепсии, проявляющийся единственным видом приступов – первично-генерализованными тонико-клоническими судорожными пароксизмами при отсутствии ауры и четкого фокуса на ЭЭГ. Дебют заболевания наблюдается в очень широком возрастном диапазоне: от 1 до 30 лет с максимумом в пубертатном периоде (средний – 13.5 лет). Клинически ГСП проявляются внезапным (без ауры) выключением сознания с падением пациентов, судорогами, заведением глазных яблок, расширением зрачков. Сначала наступает короткая тоническая фаза, переходящая в более длительную клоническую с последующим постприступным оглушением. Продолжительность ГСП составляет от 30 сек до 10 мин (в среднем 3 мин). Частота приступов невысока – от единичных в год до 1 раза в месяц, без тенденции к серийному и статусному течению. Чрезвычайно характерна приуроченность большинства приступов к периоду пробуждения и, реже, засыпания. Наиболее значимый провоцирующий фактор – депривация сна и внезапное насильственное пробуждение. Возможно учащение приступов в перименструальном периоде. ЭЭГ-исследование в межприступном периоде у половины больных может быть в пределах нормы. Характерна генерализованная пик-волновая активность с частотой от 3 Гц и выше, нередко с амплитудной асимметрией или бифронтальным преобладанием. Могут выявляться различные региональные паттерны. Лечение. Ремиссия достигается у 75-80% больных. Базовые препараты – карбамазепин и вальпроаты. При отсутствии на ЭЭГ генерализованной пик-волновой активности лечение начинается с карбамазепина, который более эффективен, чем вальпроаты. При присоединении к генерализованным судорожным приступам абсансов или миоклонических пароксизмов, или при появлении на ЭЭГ генерализованной пик-волновой активности, базовый препарат – только вальпроаты. Средняя дозировка карбамазепина – 15-25 мг/кг/сут в 3 приема; вальпроатов 20-50 мг/кг/сут в 3 приема. К резервным препаратам относятся барбитураты (фенобарбитал 1.5-3.0 мг/кг/сут в 1-2 приема; гексамидин, бензонал), гидантоины (дифенин 4-8 мг/кг/сут в 2 приема). В редких резистентных случаях возможны комбинации: карбамазепин + вальпроаты; карбамазепин + барбитураты; вальпроаты + барбитураты; барбитураты + гидантоины. При неадекватном лечении возможно присоединение к ГСП абсансов или миоклонических приступов с трансформацией в юношескую миоклоническую эпилепсию.