Opticomyelitis (Devic's disease)
Opticomyelitis (Devic's disease)
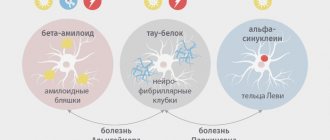
Devic's neuromyelitis optica (neuromyelitis optica, Devic's disease/syndrome) is a neurodegenerative process of an autoimmune nature, accompanied by the destruction of myelin sheaths.
Thus, this is one of the demyelinating diseases of the nervous system (more detailed information about pathology of this kind can also be found in the materials “Multiple sclerosis”, “Guillain-Barré syndrome”, “Subcortical atherosclerotic encephalopathy (Binswanger disease)”). Eugene Devic (France) was a practicing neurologist and researcher; His main publications concerned infantile chorea, cerebral glioma, tumors of the corpus callosum, and typhoid fever. In 1894, E. Devic, in collaboration with his graduate student Fernand Go, described a rare disease of the nervous system (by that time, only 16 cases were known in Europe and North America, to which Devic and Go added their own, seventeenth observation), in many ways similar to multiple sclerosis, but with a distinct specificity, namely almost isolated damage to the optic nerve and spinal cord; Such selectivity is not typical for demyelinating diseases.
It should be noted that descriptions of individual symptoms of neuromyelitis optica appeared long before the work of Devika-Go, back at the beginning of the 19th century. The outstanding English doctor and public figure, Sir Thomas Clifford Allbot, also contributed in 1870 and attracted the attention of the medical community to this specific disorder, but a detailed clinical description of neuromyelitis optica as a separate, independent disease belongs to E. Devic.
Throughout the twentieth century, despite a number of distinctive features, Devic's opticomyelitis was considered one of the atypically severe variants of multiple sclerosis, and some sources stubbornly continue to reproduce this hopelessly outdated interpretation. In the 2000s, mainly through the work of the research sector of the world-famous Mayo Clinic (Rochester, USA), this uncertainty was finally eliminated: it was possible to identify a humoral mechanism that is aggressive towards perivascular protein, and to find reliable biochemical markers of neuromyelitis optica (immunoglobulin antibodies), which do not occur in multiple sclerosis. According to the Mayo Clinic, this was the first time a specific molecular target for the autoimmune-inflammatory demyelinating process had been discovered. As of today, the presence of six types of antibodies is assumed, three of which have already been discovered.
Thus, Devic's neuromyelitis optica is indeed an independent nosological entity, and not a variant of multiple sclerosis.
Epidemiological estimates are difficult due to the relative rarity of this syndrome and the likely occurrence of numerous diagnostic errors. It is reported that "true" Devic's disease occurs approximately fifty times less frequently than classic multiple sclerosis, accounting for 1-2% of the total demyelinating diseases. The age range for initial diagnosis ranges from one year to 77 years; Most often, manifestation occurs in the period of 35-45 years. Women get sick much more often than men (3-9 times according to various sources). Between half and two-thirds of patients with Devic's neuromyelitis optica also have another autoimmune disorder.
Devic's optomyelitis
Neuromyelitis optica (Devic's disease) has long been controversial. Should this disease be considered a subtype of multiple sclerosis (MS) or an independent nosological form? The discovery of specific antibodies to aquaporin-4 (AQP4) channels, which are widely present in astrocyte processes, allowed the definitive separation of these diseases. In 2006, the criteria for neuromyelitis optica spectrum disorders (NMOSD) were revised to require detection of antibodies to AQP4 for diagnosis. These criteria and the McDonald criteria used to diagnose MS emphasize neuroimaging findings. However, it often happens that MRI results do not differentiate between these conditions, for this reason NMOSD is often mistaken for MS, which can lead to iatrogenicity, since NMOSD requires long-term immunosuppressive therapy to prevent relapses, and therapy for MS includes, among other things, natalizumab and interferon-β, which can lead to exacerbation of NMOSD. [2]
Neuromyelitis optica typically manifests as optic neuritis and longitudinally extensive transverse myelitis (LETM), although recent studies suggest involvement of the circumventricular organs and skeletal muscles. Neuromyelitis optica occurs with relapses, is often associated with other autoimmune diseases and is sometimes a component of paraneoplastic syndrome. The specificity of testing antibodies to AQP4 approaches 100%. Clinical, immunohistopathological and in vitro physiological data support that these autoantibodies are key in the pathogenesis of neuromyelitis optica. Recent animal studies provide limited histopathological characteristics in the absence of clinical deficits. The recently described antibodies to myelin oligodendrogliocyte glycoprotein in a small group of patients with the NMOSD phenotype and the absence of AQP4-IgG provide grounds for further identification of new nosological forms.
Devic's neuromyelitis optica is an inflammatory autoimmune disease of the nervous system that manifests as longitudinal extended transverse myelitis (LETM) and optic neuritis. The discovery of serum immunoglobulin G specific to aquaporin-4 channels made it possible to unify neuromyelitis optica itself and the spectrum of disorders associated with it (NMOSD), as well as to reliably distinguish it from MS. However, the relationship between idiopathic neuroneuritis optica and transverse myelitis within AQP4-IgG-seronegative neuromyelitis optica with the presence of alternative autoantibodies, for which CNS involvement has not yet been determined by immunohistopathological methods, remains unclear.
NMOSD has not been assessed by race or ethnicity in different populations, but in Western countries there is a disproportionate distribution among non-white patients. In Asia, NMOSD is diagnosed slightly more frequently than MS. Women get sick more often, according to various estimates, from 3.6:1 to 10.4:1.9. The disease manifests itself at the age of 35–45 years, but 18% of cases occur in children or the elderly. Although most cases are sporadic, rare cases of familial seropositive AQP4-IgG neuromyelitis optica with a classic phenotype indicate a genetic predisposition.
Clinic
Table 1 summarizes the 2006 criteria for neuromyelitis optica. Devic in 1894 first described a woman with urinary retention, paraplegia, bilateral blindness, and papilledema. Autopsy revealed acute myelitis and bilateral optic neuritis. Simultaneous bilateral neuroneuritis optica and transverse myelitis is a rare modern clinical picture of neuromyelitis optica. Such cases are usually monophasic, occur equally often in men and women, and are AQP4-IgG seronegative. Thus, they cannot be identical to AQP4 autoimmune channelopathy. A longer interval between initial clinical symptoms and late manifestations, manifestation at an older age and female gender predict a recurrent course of neuromyelitis optica. Although, unlike MS, a secondary progressive course is not typical.
Unilateral or bilateral optic neuritis is more severe in NMOSD than in MS, with complete recovery in only 32% of cases; transverse myelitis (usually longitudinally distributed) with complete clinical recovery - only in 17% of cases (Fig. 1). Small lesions in the spinal cord are detected on MRI in 14% of cases at presentation, and in case of relapse, 92% of lesions are already longitudinally widespread. Damage to the conus medullaris and lumbosacral regions is not typical.
Table 1 ❘ Criteria for opticomyelitis Devika, 2006
Figure 1 ❘ Longitudinal extended transverse myelitis (LETM) of the cervical (Image A, T2-WI) and cervicothoracic (Image B, T2-WI) spine with inhomogeneous contrast enhancement (Image C, post-contrast T1-WI) in AQP4-IgG-positive patients. [1]
Figure 2 ❘ T2-weighted MRI of a 36-year-old woman with LETM.
The lesion extends beyond the spinal cord (dashed line). [3] Neuromyelitis Optica Spectrum Disorders (NMOSD)
The presence of AQP4-IgG unites patients with minor forms of neuromyelitis optica who do not fully meet the diagnostic criteria. Clinical manifestations essentially repeat the symptoms of neuromyelitis optica: neuritis optica, LETM, damage to the circumventricular organs and autoimmune AQP4 myopathy (Table 2). Neuromyelitis optica and NMOSD can develop in cancer (4–5% of all NMOSD cases and 15% in older patients) in the form of paraneoplastic syndrome. However, the presence of AQP4-IgG alone without clinical manifestations of central nervous system involvement does not allow the diagnosis of NMOSD.
Table 2 ❘ Spectrum of neuromyelitis optica-associated disorders (NMOSD), defined as AQP4-IgG seropositive status (2015 data).
Brain damage
Involvement of the brain in the pathological process, according to MRI, occurs in 60% of cases during the first attack, while in 10% of cases the pathology may resemble that of MS. Another 10% of patients have a typical location of lesions for neuromyelitis optica (hypothalamus, periventricular areas) (Fig. 3). The disease may be asymptomatic or have clinical manifestations of damage to the circumventricular organs: uncontrollable nausea and vomiting (area postrema and the bottom of the fourth ventricle), hiccups, syndrome of inappropriate secretion of antidiuretic hormone, narcolepsy or anorexia (hypothalamus) or endocrinopathies of the posterior pituitary gland. The brain stem is affected in 31% of cases (more common in non-white patients), resulting in vomiting (33%), hiccups (22%), oculomotor disturbances (20%) and itching (12%), and hearing loss may occur in 3.3% , facial paralysis, vestibulopathy, trigeminal neuralgia. Five women with posterior reversible encephalopathy syndrome were also reported. Clinical presentation and brain lesions in children may resemble acute disseminated encephalomyelitis (ADEM).
Figure 3 ❘ Distribution of brain lesions. Lesions are localized in areas of increased expression of aquaporin-4 (white dots in the diagram) [1]
Patient 1: localized around the 3rd ventricle and hypothalamus; Patient 2: localized around the 4th ventricle; Patient 3: localized around the 4th ventricle and aqueduct; Patient 4: periependymal localization, lesions around the lateral ventricles, lesion of the spinal cord extending to the brainstem; Patient 5: lesions of the thalamus, hypothalamus, chiasm, as well as lesions around the 4th ventricle, subpial lesions in the cerebellar hemispheres; Patient 6: localized around the 4th ventricle and cerebellar peduncles.
Concomitant autoimmune diseases
Organ-specific and non-specific autoimmune manifestations (autoimmune diseases or antibody markers) often accompany NMOSD. For example, Sjögren's syndrome and systemic lupus erythematosus. Transverse myelitis is always a manifestation of NMOSD, and not a complication of vasculitis in systemic connective tissue diseases. Autoimmune myasthenia gravis occurs 100 times more often in patients with NMOSD (2%) than in the general population (0.02%). There have also been cases of celiac disease, autoimmune gastritis, autoimmune thyroiditis, ulcerative colitis and sclerosing cholangitis.
Skeletal muscle damage
Hyperkalemia may precede or accompany NMOSD. An indicative case is with LETM and neuromyelitis optica, when symptoms of proximal myalgia (CK >10,000 U/L) appeared in response to corticosteroid therapy. A biopsy of the affected muscle showed sarcolemmal loss of AQP4, as well as linear deposits of IgG and complement proteins, and the muscle showed the histological pattern characteristic of CNS lesions: lymphohistiocytic infiltrates and some eosinophils. Healthy muscles have a large number of AQP4 channels on their surface, and in necrotizing autoimmune myopathies and dermatomyositis, partial loss of AQP4 also occurs, but without deposition of immune complexes in the sarcolemma.
Myelin oligodendrogliocyte glycoprotein (MOG)
Antibodies to myelin oligodendrogliocytic glycoprotein (MOG-IgG) were previously considered marker antibodies for childhood MS; today they are found in children with ADEM, and sometimes in adults and children in cases of relapse of AQP4-IgG-positive and AQP4-IgG-negative neuromyelitis optica. MOG-IgG-positive cases are most often monophasic, men are more often affected, the age of manifestation is younger, deep gray matter lesions are more often seen on MRI, and the prognosis of the disease is more favorable. It should be noted that these cases were not studied using immunohistochemical methods.
Patients with established diagnoses of neuromyelitis optica (MOG-IgG-positive and AQP4-IgG-negative phenotypes) had high levels of myelin basic protein in the cerebrospinal fluid in the absence of glial fibrillary acidic protein (GFAP), which favors a demyelinating process rather than astrocytopathy.
Until morphological data are available for MOG-IgG-positive cases, it is more correct to call them “MOG-autoimmune optic neuritis, transverse myelitis or encephalomyelitis” rather than NMOSD.
Laboratory and instrumental research
MR features
Neuroimaging of transverse myelitis usually reveals spinal cord lesions extending over 3 vertebrae, but in 14% of cases the lesion may be shorter at onset. Extended lesions are edematous, accumulate contrast unevenly, and sometimes there are areas of central necrosis or cavities. Later, focal spinal cord atrophy can be observed. Damage to the central gray matter helps distinguish this pathology from demyelinating diseases. DTI (diffusion tensor imaging) reveals in patients within a year after the last exacerbation more pronounced radial diffusion in the white matter than in MS, which corresponds to greater tissue destruction.
Features of optic neuritis in neuromyelitis optica include the bilateral and extended nature of the lesion, damage to the chiasm and optic tracts. Optical coherence tomography and visual evoked potentials complement and complete instrumental diagnostics. In the brain, lesions of the hypothalamus and periventricular region are most characteristic, but in 10% of cases an MS-like picture can be observed, which meets the Barkoff criteria. [1]
The characteristic imaging features of NMOSD and MS were studied (Tables 3,4,5). Two experienced neuroradiologists, blinded to clinical examination and diagnosis, assessed each lesion on MRI. This study found that the distribution of lesions differed significantly between NMOSD and MS. Although round lesions/Dawson's fingers and isolated U-fiber/juxtacortical white matter lesions were found to have a significantly higher incidence in MS than in NMOSD compared with previous European studies, the incidence of this type of lesion in NMOSD in this study (round lesions 21.5%, isolated lesions of U-fibers 10.1%) was higher. Past studies have shown that the presence of such lesions can discriminate between MS and NMOSD with high sensitivity and specificity, although this is controversial in the Asian population with NMOSD. Genetic or environmental factors have been reported to influence imaging differences in MS patients across different domains. Imaging features of NMOSD may also differ between Asian and European patients.
Although cloud-like enhancement is a characteristic enhancement pattern of NMOSD, no patients with NMOSD in this study exhibited this feature. Cloud-like contrast enhancement is defined as “multiple patchy enhancement with blurred edges in adjacent areas compared with isolated enhancement of the lesion.” However, the diagnostic threshold for this trait may vary slightly depending on the evaluator, which may affect the results of the study.
Although bilateral optic neuritis and chiasmal involvement are specific features of NMOSD, no significant differences were found between NMOSD and MS in this study. The results can be explained by the following: first, patients were recruited in both acute and chronic phases, whereas previous studies only included patients in the acute phase; second, this retrospective analysis of optic nerve lesions included different numbers of patients with NMOSD and MS. Thus, further prospective studies with clear eligibility criteria are required. [2]
Table 3 ❘ Characteristics of brain lesions in MS and NMOSD. Median values are shown. [2] *dirty lesions are defined as lesions with signal intensity intermediate between those of focal lesions and normal-appearing white matter
Table 4 ❘ Characteristics of spinal cord lesions in MS and NMOSD. Median values are shown. [2]
Table 5 ❘ Characteristics of lesions of the optic nerve in MS and NMOSD.
Median values are shown. [2] CSF changes
Changes in the cerebrospinal fluid in neuromyelitis optica are quite different from those in MS. Only 16% of AQP4-IgG-positive neuromyelitis optica have oligoclonal immunoglobulins, 50% have pleiocytosis: on average about 19/μl of leukocytes, and in 6.2% it reaches 100/μl, mainly due to neutrophils, basophils and eosinophils. Levels of GFAP, a marker of astrocyte damage, are higher in neuromyelitis optica than in MS and other neurological diseases, while the cytoplasmic protein S100B, which is found predominantly in astrocytes, increases moderately. Myelin basic protein and neurofilament heavy chains showed similar levels across the three diseases. GFAP concentrations normalize almost immediately after initiation of therapy, while myelin basic protein levels remain high, likely marking demyelination. High levels of myelin basic protein, S100B and GFAP during exacerbation correlate with the extent of LETM and worsen EDSS (Expanded Disability Status Scale) scores. The values of this scale after 6 months correlate only with the level of GFAP during an exacerbation.
Antibody profile
A huge number of autoantibodies (organ-specific and non-specific) help to distinguish between neuromyelitis optica and MS, while titers are generally low (Table 6). Antibodies to aquaporin-1 channels have been reported, but their clinical utility has not yet been assessed.
Table 6 ❘ Antimyelitis optica antibody profile [1]
Prognosis
Before the discovery of AQP4-IgG, the prognosis of neuromyelitis optica was extremely unfavorable, since therapeutic options were not defined. In a review of case series, Wingerchuk et al. 1999, it was reported that when neuromyelitis optica manifests itself, the development of the main symptoms occurs quickly, on average within 5 days, followed by moderate recovery. Subsequently (in case of relapse), the interval between the initial LETM and optic neuritis increases to an average of 166 days, severe repeated attacks are observed within 3 years, and disability occurs in stages. A third of patients die from respiratory complications.
According to the results of a 2012 study of AQP4-IgG-positive patients from the UK and Japan, permanent bilateral visual impairment develops in 18% of patients (the average duration of the disease is 75 months), permanent motor impairment (inability to walk more than 100 meters without assistance) - in 34%, 23% confined to a wheelchair. With an average disease duration of about 99 months, 9% of patients die after 4.5 attacks. In an English cohort study, African-Caribbean patients had an earlier age of onset and a higher likelihood of developing visual impairment than white patients.
According to a 2013 report that examined AQP4-IgG-positive and AQP4-IgG-negative patients, 65% of patients lost vision an average of 8.3 years after disease onset (50% monolateral, 15% bilateral), and more than a third could not move without assistance.
Data suggest a more favorable prognosis for the disease after the discovery of AQP4-IgG, presumably due to the possibility of early diagnosis and, as a consequence, early initiation of treatment aimed at reducing the level of immunoglobulins, blocking the activation of the complement system and the action of interleukin-6.
Treatment
Therapy for neuromyelitis optica has two goals: relief of the acute condition (in order to minimize neurological deficits and increase the chance of recovery) and prevention of relapses leading to disability. Therapeutic decisions should be made individually based on the clinical picture and should not be based on AQP4-IgG levels. To date, there are no prospective controlled studies evaluating treatment options for neuromyelitis optica (Table 7).
Table 7 ❘ Therapy for neuromyelitis optica [1]
As for other IgG-mediated autoimmune diseases, intravenous methylprednisolone (IVMPD) and plasmapheresis are considered optimal first-line treatment options. It is recommended to use 1 g of IVMPD for 3 to 5 days. Oral forms of prednisolone are used to prevent early relapse with subsequent transition to non-steroidal immunosuppressants. In case of severe exacerbations, IVMPD alone may not be enough; adding plasmapheresis to therapy (from 5 to 7 sessions) improves the results of therapy. In some particularly severe cases, cyclophosphamide (750–1000 mg/m2) was more effective than pulse therapy with methylprednisolone.
Intravenous immunoglobulins (IVIG) with or without plasmapheresis showed good results in a study of 10 patients who did not receive IVMPD.
Further research is needed to determine optimal therapeutic options based on patient age, comorbidities, and seizure severity.
Relapse Prevention
Immunomodulatory therapies used for MS are not effective for NMOSD, and some medications may cause worsening. Retrospectively, using available data from a large number of patients, a number of drugs have been shown to be effective: azathioprine, mycophenolate mofetil and rituximab.
Azathioprine (at least 2 mg/kg/day) reduced annual relapse rates in a study of 99 patients. Blood counts, liver and kidney function should be monitored throughout treatment, and it is also advisable to determine the activity of thiopurine methyltransferase (the enzyme responsible for the activation of azathioprine) before starting treatment. Since the effect of azathioprine is delayed, oral forms of glucocorticosteroids are used in the first few months. In a study of 32 patients, azathioprine reduced relapse rates by 72% but was ineffective in half the patients despite combination with prednisolone.
Mycophenolate mofetil may also be used for long-term immunosuppression. In a study of 28 patients, the relapse rate was reduced by 87%.
Rituximab, an anti-CD20 monoclonal B-cell inhibitory agent, has shown high efficacy in the treatment of NMOSD. Open-label studies using different dosing protocols and monitoring of CD19+ lymphocytes suggest that the drug should be restarted when B cells reappear in the blood. The annual relapse rate is reduced to 87%, and 60–70% of patients are relapse-free over the next 3 years.
Eculizumab, a monoclonal drug that binds to the complement protein C5, showed no relapses at one year in 12 of 14 patients in a pilot open-label study. During treatment, the median relapse rate was reduced from 3 to 0, and the EDSS score improved from 4.3 to 3.5.
Tocilizumab, an anti-IL-6 receptor monoclonal drug, showed efficacy in an open-label study in 7 patients. After one year, the relapse rate decreased from 2.96 to 0.4. EDSS scores were significantly reduced, and neuropathic pain and general weakness were reported less frequently by patients.
Oral corticosteroids as monotherapy or adjunctive therapy have been shown to prevent relapses in a small retrospective study. Combination therapy minimizes the side effects of extended corticosteroid therapy. During long-term treatment, it is recommended to prevent osteoporosis and monitor blood glucose.
Cyclosporine A is used in combination with oral corticosteroids. The annual relapse rate in the 9 treated patients decreased from 2.7 to 0.38.
In a retrospective study of 14 patients, methotrexate showed a significant reduction in relapse rates (0.18 during therapy and 1.39 before), 43% of patients were free of relapses, and the rate of disability stabilized or decreased by 79%.
Cyclophosphamide reduced the median EDSS from 8 to 5.75 in 4 patients with LETM.
Autologous bone marrow transplantation stopped disease progression in 3 of 16 patients who were resistant to previous treatment. These patients stopped receiving any therapy (median 47 months). The remaining 13 patients required continued therapy due to deterioration of general indicators or relapses. Allogeneic bone marrow transplantation was effective in 2 patients with aggressive neuromyelitis optica in whom previous therapy, including autologous bone marrow transplantation, had failed. Both patients were relapse-free at 36 and 48 months. Pathogenesis
The beneficial effect of plasmapheresis and drugs that suppress B-lymphocyte activity suggests that AQP4-IgG plays a major role in pathogenesis.
Immunopathogenesis
Around the hyalinized vessels, complement activation products are detected, which bind to IgG molecules. The predominance of polymorphonuclear proinflammatory cells is consistent with the participation of IgG and complement in the pathogenesis of neuromyelitis optica. Loss of aquaporin-4 channels by astrocytes surviving sublytic CNS lesions distinguishes neuromyelitis optica from MS and may indicate potentially reversible histopathological changes. Some lesions show significant inflammation and astrocyte loss of AQP4, but retain GFAP. Other lesions show low astrocytic immunoreactivity for GFAP with relatively intact myelin. These data suggest that loss of AQP4 causes astrocyte damage, ultimately leading to secondary demyelination. Subsequently, extensive demyelination, cavitation, necrosis, and axonal loss (spheroidosis) are noted in the gray and white matter of the spinal cord.
Role of AQP4-IgG
The appearance of IgG in response to protein autoantigens reflects the interaction of regulatory and helper T lymphocytes with antigen-specific B lymphocytes. The role of AQP4-specific T lymphocytes is unclear, but they may contribute to damage to the blood-brain barrier. IgG-producing plasma cells were found in the studied lesions of neuromyelitis optica. More and more information is emerging about the intrathecal synthesis of AQP4-IgG, the high level of which in the cerebrospinal fluid significantly correlates with the severity of the exacerbation. An increased level of IL-6 is detected in the blood plasma, which promotes the survival of lymphocytes.
To trigger the pathological process, IgG binding to the extracellular domain of AQP4 is required. AQP4-IgG are predominantly Ig of the G1 subclass - a potential activator of the complement system. In the astrocyte membrane, AQP4 exists as a tetramer and has two isoforms: full (M1) and short (M23).
M23-AQP4 spontaneously forms orthogonal arrays of particles, while most homomeric M1 tetramers remain singlets.
When exposed to AQP4-IgG, astrocytes rapidly internalize M1 tetramers, while binding of M23 causes their fusion into larger arrays that exceed the ability of astrocytes to endocytose. When complement becomes available, large arrays of AQP4, which contain tightly packed IgG1 Fc tails, avalanche-like activate the complement system, promoting the recruitment of leukocytes, increasing vascular permeability and massive sweating of plasma rich in IgG, complement system proteins, which, when bound to the antigen- the antibody will enhance complement activation.
In vitro studies revealed a relationship between the severity of an attack of neuromyelitis optica and the degree of complement-mediated damage to cells expressing AQP4 due to exposure to IgG in patients with neuromyelitis optica (6 patients with severe exacerbation, 6 patients with mild exacerbation, p = 0.05). Degranulation of NK cells, which are activated in a complement-independent pathway by IgG binding to AQP4 (antibody-dependent cellular cytotoxicity), also causes astrocyte death in vitro.
Blocking water flow by antibodies plausibly explains the visible myelin swelling in early lesions of neuromyelitis optica, which is a possible precursor to demyelination. Antibodies that have the potential to block water flow independent of AQP4 lesion (i.e., temperature independent) have demonstrated this effect in clawed frogs (Xenopus). These antibodies were obtained from at least 50 patients with neuromyelitis optica and high levels of AQP4-IgG. Thus, data were obtained that dispute the major role of AQP4-IgG, although it is worth considering that they were based on the study of monoclonal AQP4-IgG and sera from a relatively small number of individual patients using quantum dot technology.
Another property of the AQP4-IgG complex is disruption of glutamate homeostasis. The excitatory amino acid transporter EAAT2 is non-covalently associated with aquaporin channels on the presynaptic membrane of astrocytes. EAAT2 accounts for up to 90% of utilized glutamate in the CNS; in addition, this transporter can be taken up by AQP4-IgG. The use of these antibodies on astrocyte cell cultures led to a decrease in the functional uptake of glutamate. Moreover, in subacute spinal cord lesions in the central gray matter, severe EAAT2 deficiency is observed. Thus, the particular vulnerability of oligodendrocytes to glutamate toxicity is another plausible explanation for demyelination in neuromyelitis optica.
Animal models
Animal models described to date recapitulate some of the histopathological characteristics of neuromyelitis optica, but not the clinical findings. The lesions characteristic of neuromyelitis optica do not occur when serum IgG from affected patients is injected into rats with an intact blood-brain barrier. However, when it is used in rats with experimental autoimmune encephalitis, they develop the full range of expected pathophysiological changes. Also, IgG did not cause any changes in young rats, in which the content of endothelial, i.e., barrier, cells in the central nervous system is extremely low.
On the other hand, astrocyte damage and loss of AQP4 in the spinal cord have been reported following IgG administration to mice pretreated with complete Freund's adjuvant with or without pertussis toxin. The authors suggested that the mycobacterial adjuvant helps to increase the permeability of the blood-brain barrier. Focal lesions characteristic of neuromyelitis optica developed in mice injected with IgG directly into the cerebral hemispheres, but only if human plasma was also injected as a source of complement. [1]
Sources
1. Zekeridou A., Lennon VA Aquaporin-4 autoimmunity //Neurology-Neuroimmunology Neuroinflammation. – 2015.
2. Tatekawa H. et al. Imaging Differences between Neuromyelitis Optica Spectrum Disorders and Multiple Sclerosis: A Multi-Institutional Study in Japan //American Journal of Neuroradiology. – 2021.
3. Chee CG et al. MRI Features of Aquaporin-4 Antibody–Positive Longitudinally Extensive Transverse Myelitis: Insights into the Diagnosis of Neuromyelitis Optica Spectrum Disorders //American Journal of Neuroradiology. – 2021.
1.What is Devic's syndrome (opticomyelitis)
Devic's syndrome is a special form of multiple sclerosis, characterized by a rapid course and severe symptoms. Pathogenetically, neuromyelitis optica belongs to the group of autoimmune diseases. The neurological manifestations of Devic's syndrome are caused by a combination of optic neuritis and damage to the protective fat-like membrane of the spinal cord (transverse myelitis). Developing symptoms affect vision, motor functions, as well as processes regulated by the autonomic nervous system.
Devic's syndrome predominantly develops in old age, and women are more often affected. Foci of transverse myelitis are usually localized at the level of the thoracic or cervical segment of the spinal cord. Vision may decrease symmetrically or in only one eye. The degree and speed of its loss varies from person to person, up to complete blindness. Severe clinical manifestations also include decreased sensitivity, leading to dysfunction of the intestines and bladder.
The reasons for the development of Devic's syndrome are not clear. There is some connection with past infectious diseases - in most patients, the first manifestations of the disease were preceded by fever and other phenomena characteristic of a viral or bacterial infection. It is difficult to say whether they were a true manifestation of a possible immune reaction to pathogenic flora or whether they represented a prodromal period of neuromyelitis optica itself.
A must read! Help with treatment and hospitalization!
Difference from multiple sclerosis
Multiple sclerosis and neuromyelitis optica are similar diseases. For a long time, until 2004, isolated pathology of the optic nerve and spinal cord was classified as a separate form of MS. The pathogenesis of the disease is general. In both cases, there is a disturbance in the structure of the pathways with the progression of characteristic symptoms.
In 2004, scientists isolated specific NMO-IgG antibodies from the blood of patients suffering from Devic's disease. It turned out that the presence of the corresponding immunoglobulins is not typical for patients with multiple sclerosis. This fact became fundamental for identifying neuromyelitis optica as an independent disease.
The following symptoms also remain characteristic of multiple sclerosis:
- Decreased muscle strength in all four limbs.
- Intellectual impairment.
- Emotional instability. Euphoria alternates with bouts of depression.
- Itching, burning and tingling sensation in the fingers and toes.
Differential diagnosis using MRI is not always reliable, however, it is widely used in practice.