History of discovery
British ophthalmologist Warner Tay and American neurologist Bernard Sachs independently described the disease in 1887 and developed diagnostic criteria to distinguish the disease from other neurological disorders with similar symptoms.
Bernard Sachs was the first to hypothesize that this pathology is genetic. His intuitive assumption was confirmed in the mid-twentieth century after the rediscovery of Mendel's laws.
Bernard Sachs proposed a name for the new disease that can also be found in modern medical literature - amaurotic familial idiocy.
Causes of the disease
For a long time, doctors could not answer the question of what causes Tay-Sachs disease. The causes of the pathology became known only in the middle of the twentieth century, when ideas about genetics were formed. Studies have shown that the disease develops as a result of a mutation in the HEXA gene, which is located on the 15th chromosome. The disease is a type of GM2 gangliosidosis, a genetic pathology associated with a deficiency or decreased activity of hexosaminidase. Amaurotic idiocy occurs as a result of a decrease in the activity of hexazaminidase A or a deficiency of this enzyme.
The disease is transmitted in an autosomal recessive manner, therefore if a person has a healthy HEXA gene in his genotype, then he does not develop Tay-Sachs disease. The genetics of the disease is similar to the inheritance of such pathologies as Gaucher disease, Urbach-Wiethe disease, Dabin-Johnson syndrome: if both parents were carriers of the mutated gene, the probability of having a sick child is 0.25%, if both mother and father were sick, then both In children, the disease manifests itself in almost 100% of cases.
What is Tay Sachs disease
Tay Sachs disease is a genetic pathology that affects the brain and nervous system. This includes the spinal cord and meninges. In the first six months of a child's life, his development is normal. Then disruptions in brain activity begin. Newborn children are diagnosed with gangliosidosis gm1. Such patients die after 3 - 4 years. This pathology has a second name – gm2 gangliosidosis.
- Etiology
- Classification
- Symptoms
- Diagnostics
- Treatment
- Prevention
Main forms of the disease
It is customary to distinguish three main forms of the disease. The most common of them is infantile. Children with Tay-Sachs disease develop normally until 6-7 months. After this, a slow but irreversible process of decline in mental and physical abilities begins.
There is also a juvenile form of the disease. Compared to infants, it is less common. Until approximately 3-10 years of age, the child develops in the same way as his peers, but over time, a slow decline in cognitive and motor functions begins, dysarthria, dysphagia, and ataxia develop.
Late-onset Tay-Sachs disease is the rarest form of the disease. The first signs of the disease usually appear after 30 years. However, cases of earlier onset of symptoms (15-18 years) have also been recorded. This form of the disease has the most favorable prognosis, since its progression can be stopped.
Affected Populations
Tay-Sachs disease affects men and women equally. The disease is more common among Jews of Ashkenazi origin, that is, people from Eastern or Central Europe. About one in 30 Ashkenazi Jews carries the altered Tay-Sachs disease gene. Additionally, one in every 300 people of non-Ashkenazi Jewish background is a carrier.
In this specific Jewish population, about 1 in 3,600 live births are affected. The disease is also seen in some people of Italian, Irish Catholic, and non-Jewish French-Canadian ancestry, especially those living in the Cajun community of Louisiana and southeastern Quebec. In the general population, the rate of transmission of the altered gene is approximately 1 in 250-300 people.
The late form of Tay-Sachs disease is less common than the infantile form. However, rare diseases such as late-onset Tay-Sachs disease often go unrecognized. These disorders are underdiagnosed, making it difficult to determine the true incidence of such disorders in the general population.
Symptoms
Regardless of the form of the disease, several main symptoms appear: dysphagia, ataxia, loss of cognitive function, muscle atrophy. If a child under one year of age reacts sharply to sharp sounds, gains weight poorly and cannot relax his muscles, parents should show him to specialists - this is how Tay-Sachs disease begins in infants. Symptoms become increasingly severe. After 6 months, motor activity decreases, the baby loses the ability to sit independently and change position. Blindness gradually develops, hearing decreases, muscles atrophy and complete paralysis of the body develops.
In the juvenile form, in addition to the main symptoms, dysarthria (decreased speech clarity), spasticity, and impaired coordination of movements are observed. There is a gradual loss of cognitive functions - a decrease in memory, attention, and performance. Dementia develops. In the later stages of the disease, seizures appear.
The first symptoms of the adult form of the disease are difficulty swallowing, loss of coordination and dysarthria. Mental disorders similar to the symptoms of schizophrenia (visual and auditory hallucinations, apathy, decreased emotionality) often appear. Without treatment, cognitive function deteriorates. Only for this form of the disease there is an effective treatment that can stop Tay-Sachs disease. The National Guide to Neonatology says that effective methods for diagnosing the adult form of the disease appeared only in the 70s; before that, the disease was considered a childhood disease.
Tay-Sachs disease (Gangliosidosis GM2, Tay-Sachs idiocy, Early childhood amaurotic idiocy)
The clinical picture of the disease reflects the processes of damage to the central nervous system. In the infantile form, the first symptoms become noticeable by 3-5 months; before this, development corresponds to the norm: the child holds his head, rolls over onto his stomach and back, coos, smiles when he sees an adult, and makes visual contact. By 6 months, interest in the outside world decreases. The baby looks to the side for a long time, is apathetic, inactive, sensitive to loud sounds and bright light. He stops recognizing close people and has difficulty focusing his eyes on his favorite toys.
After six months, the delay in mental and physical development becomes even more noticeable. Muscular hypotonia increases, previously acquired skills are lost: holding the head and body in an upright position, turning over while lying on a horizontal surface, sitting (attempting to sit), grasping a toy and transferring it from hand to hand. By 8-10 months, start reflexes—reactions to sudden sound, light, tactile and olfactory stimuli—intensify. Interest in the events happening around almost completely disappears. By 12 months, the ability to swallow is impaired, hearing and vision are noticeably reduced, and the breathing cycle becomes difficult. Muscles undergo atrophy, paralysis and convulsions develop in the form of tonic-clonic generalized and partial seizures. In the second year of life, phenomena of decerebrate rigidity and bulbar-pseudobulbar syndrome occur.
The juvenile form debuts with less obvious symptoms. At the initial stage, emotional instability somewhat intensifies; when performing complex motor complexes - running, walking, fast writing - a barely noticeable incoordination of movements appears. After some time, clumsiness and awkwardness increase and are noticed by others and by the child himself. By adolescence, the gait becomes shaky and unstable. Hyperkinesis is formed - sudden involuntary movements of various muscle groups. Coordination problems make it difficult to continue schooling. At the same time, speech disorders of a complex cerebellar-dysarthric nature appear: smoothness and rhythm are lost, pronunciation becomes slow, blurred, and slurred. Late stages of the disease are characterized by frequent epileptic seizures, a persistent decrease in intellectual functions (dementia), loss of voluntary movements, and paralysis.
The chronic form of the disease has less pronounced symptoms and a relatively mild course. Patients experience mood swings, clumsiness, and poor pronunciation. Over the course of several years, intellectual functions have been declining: the ability for abstract thinking, comparison and analysis of phenomena and objects is lost, forgetfulness and absent-mindedness increase. Several years after the onset of the disease, mental disorders develop: patients have inadequate behavior, are affectively excitable, prone to states of agitation and deep depression, and are susceptible to psychosis with hallucinations and delusions. Over a long period of time, organic dementia develops.
Establishing diagnosis
Doctors are not always able to make the correct diagnosis when it comes to such a rare pathology as Tay-Sachs disease. The symptoms, genetics and treatment of the disease are being actively studied by specialists. Regardless of the form of the disease, there are several diagnostic procedures that are performed if its presence is suspected. One of them is the determination of the activity of the enzyme hexosaminidase in blood serum, leukocytes or fibroblasts. In patients with Tay-Sachs disease, the activity of hexosaminidase B is always below normal, the enzyme hexosaminidase A is practically absent or its activity is significantly below normal.
Another important diagnostic criterion is the presence of a bright red spot on the cornea of the eye, which can be easily noticed by a therapist or ophthalmologist using an ophthalmoscope. A red spot on the cornea was found in all patients, regardless of age.
Unlike other lysosomal storage diseases (Gaucher disease, Standhoff syndrome, Niemann-Pick disease), Tay-Sachs disease does not cause enlargement of the liver and spleen (hepatosplenomegaly).
Diagnosis of GM2 gangliosidosis
Making a diagnosis begins with a general examination of the patient, collecting his complaints, life history in the case of a child, this information is obtained from the parents. In order to confirm the pathology, additional methods for diagnosing Tay-Sachs disease are needed - these are:
- ophthalmoscopy - studies of the structures of the eyeball; with this pathology, a red spot is noted on the retina, which is nothing more than an accumulation of gangliosides;
- determination of biochemical blood parameters - elevated lipid levels can be detected in the blood;
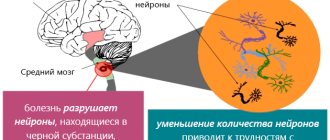
- microscopy of nerve cells - during the study, swelling and an increase in the size of neurons, accumulation of excess amounts of gangliosides in them are noticeable;
- genetic research - determination of defects in the human genetic code.
Treatment
There are currently no drugs to cure Tay-Sachs disease. The symptoms and treatment of the disease are still the subject of scientific research.
The infant form of Tay-Sachs disease is the most dangerous. If a sick child cannot swallow on his own, it is recommended to resort to artificial nutrition; it is impossible to restore physical skills. There are no drugs that can stop or reverse the development of the disease, despite all the efforts of scientists. Sick babies, even if they receive the best care, rarely survive past the age of four.
In the juvenile form of the disease, it is important that the child is constantly under medical supervision. Following the instructions of a specialist and undergoing all necessary medical procedures helps to extend the life of a sick child to 12-16 years.
The adult form of the disease progresses more slowly than others and is often treatable. For mental disorders, patients are prescribed lithium or cesium chloride. Clinical trials have shown that pyrimethamine can significantly slow down, and in rare cases completely stop, the progression of the disease by increasing the activity of hexosaminidase B.
Main symptoms
The pathology is diagnosed in babies at the age of approximately six months, since until four to five months the child develops quite normally, like all children of this age.
initial stage
The first symptoms of Tay Sachs disease are that the child loses contact with the outside world, his gaze is constantly focused in one direction, the baby does not want anything, he becomes apathetic, he has no reaction to objects, sounds, familiar faces.
He has an increased reaction exclusively to loud and sharp sounds. Even when the baby looks completely healthy, parents and loved ones often notice that the child shudders sharply with his whole body when exposed to loud auditory stimulation.
A complex of disorders manifested by increased fatigue, sleep disturbances, and mood instability are called by one term - asthenia. Hereditary Friedreich's ataxia causes serious damage to the nervous system - causes, symptoms and treatment of pathology.
Development of the disease
The second stage of the disease is regression in the child’s development: acquired skills are lost, he refuses to crawl, sit, becomes passive, toys do not cause any interest, mental retardation occurs, at the same time the size of the head increases significantly, the baby begins to lose vision, and often hearing.
At a later stage, between the first and second years of life, the baby is likely to experience seizures, manifested in the form of convulsions and paralysis.
Babies do not gain weight, but quickly lose it. With this development of the disease, the child rarely survives to the age of five.
Signs of the disease in adults
The adult form is very rare and occurs in patients between 20 and 30 years of age. It is usually not fatal.
The disease manifests itself in gait disturbances and rapid deterioration of neurological functions. An adult can live with this disease for 10 to 15 years after diagnosis.
Prenatal diagnosis
Modern research makes it possible to determine early in pregnancy whether the child has inherited the mutant HEXA gene from its parents. If both parents are carriers of the disease, it is recommended to perform a chorionic villus biopsy. This is one of the most common prenatal diagnostic procedures, the purpose of which is to identify genetic abnormalities in the fetus. It is carried out at 10-14 weeks of pregnancy. Amniocentesis also gives a clear idea of whether the child is a carrier of the mutated HEXA gene. These procedures have a risk of miscarriage of less than 1%.
In the case of artificial insemination, genetic abnormalities of the fetus can be detected even before it is implanted in the uterus. For this purpose, preimplantation genetic diagnosis is performed, an analogue of prenatal diagnosis. Its main advantage is that the procedure is non-invasive and absolutely safe. It is possible to select only healthy embryos for implantation, thereby reducing the risk of having a child with Tay-Sachs disease to almost zero.
Treatment of the disease
No effective therapy for amaurotic idiocy has been found. Despite doctors' best efforts to delay death, the progressive course of the disease increases the number of deaths in children with Tay-Sachs disease under the age of five.
Treatment of Tay-Sachs disease is based on palliative care - it is necessary to alleviate the symptoms of the pathology and improve the patient’s quality of life. In the case of this disease, the following measures are used:
- tube feeding as children lose swallowing reflexes;
- antibacterial therapy to prevent the addition of pathogenic microflora;
- antiviral drugs to prevent the occurrence of viral diseases;
- immunostimulating medications are used to enhance immune defense mechanisms.