Clinical forms and approaches to the treatment of hepatic encephalopathy in patients with cirrhosis
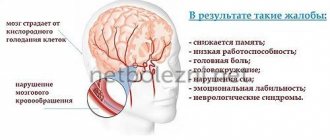
Pathogenesis Two main pathogenetic mechanisms take part in the development of PE: 1) acute or chronic liver disease, accompanied by a pronounced decrease in its detoxifying function; 2) the formation of functional or anatomical shunts between the portal and general circulatory systems, leading to the penetration of toxic products of intestinal origin into the brain. Knowledge of these mechanisms gives the doctor the opportunity, to a certain extent, to predict the development of PE in a patient with a particular pathological condition. Thus, with fulminant liver failure, PE is observed in almost all patients, after surgical application of a portacaval shunt - in 25–40%. In patients with cirrhosis, both “internal” and “external” mechanisms are involved in the pathogenesis of PE, which leads to the development of clinically manifest encephalopathy in 30–50%, and latent encephalopathy in 50–70%. Knowledge of the pathogenetic factors of PE is important for choosing the optimal treatment method (Table 1). Long-term study of the pathogenesis of PE allows us to come to the conclusion that, apparently, the considered mechanisms act in combination. The combination of unfavorable factors in a patient with acute or chronic liver pathology leads to the development of a complex of neuropsychic disorders, referred to as PE. At the same time, numerous research results give grounds to assert the role of ammonia as one of the most important neurotoxic metabolites. The most significant amounts of ammonia are formed due to deamination of amino acids in the liver; Muscles, kidneys and the digestive tract can act as additional sources. Being the main source of ammonia, the liver at the same time serves as the main site of its neutralization due to the synthesis of urea in the ornithine cycle functioning in periportal hepatocytes. A smaller portion of ammonia is involved in the synthesis of glutamine in perivenous hepatocytes, muscles and brain. Liver diseases lead to both a decrease in the rate of ammonia detoxification and an excessive entry of ammonia into the systemic circulation due to portacaval shunting. The accumulation of ammonia in the brain causes a disruption in the synthesis of astrocyte proteins, a decrease in the activity of neuronal chloride channels, and suppression of the formation of ATP and excitatory neurotransmitters - glutamate and aspartate. It is important for the clinician to know the enabling (trigger) factors, the action of which leads to the onset or worsening of the severity of PE (Table 2). Clinical forms PE is a heterogeneous group of conditions, which is divided into 5 forms (Table 3). Liver cirrhosis due to the development of portal hypertension and liver failure leads to portosystemic HE (PSE) in one of these variants, which can transform into one another. However, PSE in patients with cirrhosis should be differentiated from pseudo-PSE, described by H. Kalk in 1958 and also known as “false hepatic coma” and “electrolyte coma”. The leading role in the pathogenesis of pseudo-PSE belongs to electrolyte imbalance, represented by hypokalemia and/or hyponatremia, as well as hypomagnesemia, causing hypotonic dehydration of brain cells. The most common cause of “electrolyte coma” is an overdose of loop diuretics. Latent PSE is difficult to diagnose, as it is characterized by the absence of subjective and objective clinical symptoms, as well as changes in the recording of a spontaneous electroencephalogram (EEG). However, recognition of this form is extremely important for two reasons: • the frequency of latent PSE in patients with cirrhosis reaches 50–70%, that is, it represents the most common complication of liver diseases, regardless of etiology; • latent PSE is accompanied by an inadequate response of the patient in extreme conditions, including when driving a car, which is associated with an increased risk of creating emergency situations. For the purpose of early diagnosis of PSE, psychometric tests are widely used in clinical practice: number connections (Table 4), number-letter, lines, handwriting, arithmetic, etc. It should be taken into account that the simplicity and convenience of psychometric tests hide certain disadvantages, which include First of all, it concerns the influence of numerous exogenous and endogenous factors on their results. The likelihood of underdiagnosis of PSE can be minimized by using several tests in one patient and interpreting the results in combination. Acute and subacute recurrent forms of PSE are manifested by the clinical manifestation of encephalopathy in a patient with chronic liver disease. As a rule, such patients have a background latent PSE, which remains unrecognized in the absence of a targeted diagnostic search. Manifestation can be of varying degrees of severity - from stage I to stage IV (below) and is often, although not always, caused by the action of one or more trigger factors. Elimination of the permissive factor and therapeutic measures usually lead to the elimination of all clinical symptoms. Chronic persistent PSE is observed rarely, mainly in patients with pronounced portosystemic collaterals, including those created as a result of surgery. In addition to typical neuropsychiatric symptoms, gradually appearing symptoms of myelopathy are observed: ataxia, choreoathetosis, paraplegia, stabbing or boring pain. The damage is usually irreversible and leads to cerebral atrophy and dementia. Clinical stages Determining the stage of PE is important for assessing the patient's prognosis, since this criterion is included in the most reliable system for determining the severity of cirrhosis - the Child-Pugh functional classification. As noted, the stages of HE are essentially degrees of severity due to their potential reversibility. Symptoms characterizing PE stages 0–IV are summarized in Table 5. The so-called PE stage II–III is quite typical. flapping tremor or asterixis, manifested by rapid flexion and extension movements in the wrist joints. PE is also accompanied by EEG changes, which become apparent at stage II: flattening of the a-rhythm curve, then the appearance of t- and d-activity. In stage 0–I PE, EEG diagnosis is carried out using visual evoked potentials. To assess the state of consciousness of a patient with PE, including dynamics during therapy, the Glasgow scale can also be used (Table 6). The sum of points determines the patient's consciousness from clear (15 points) to atonic coma (3 points). The prognosis of PE is one of the most important factors determining the prognosis of liver disease. In PSE developing in patients with cirrhosis, the prognostic value is more difficult to assess than in fulminant liver failure, primarily due to the influence of numerous additional factors. However, there is statistical information in the literature that allows one to approximately determine the short-term prognosis of a patient with PSE. With stage 0–I PE during the current hospitalization, survival approaches 100%, progressively worsening with the deepening of the stage of encephalopathy: with stage II, survival is 60–75%, with III–IV – on average about 30%. 10–20% of patients survive the first episode of hepatic coma. The prognosis also depends on a number of additional factors. 1. Endogenous hepatic coma (due to loss of liver parenchyma) has a worse prognosis compared to exogenous coma (due to the release of neurotoxins of intestinal origin through portosystemic shunts). 2. The most important factor is the volume of residual liver parenchyma (indirectly determined by the severity of liver failure). 3. The prognosis may be influenced by the etiology of liver damage and the severity of collateral circulation. 4. The likelihood of an unfavorable outcome is higher in old age and in the presence of concomitant diseases. 5. The possibility and speed of elimination of trigger factors should be taken into account; First of all, this applies to stopping gastrointestinal bleeding. 6. Timely and adequate treatment of PE significantly improves the prognosis. Treatment Basic measures 1. Elimination of the etiological factor of liver disease - in cases where this is possible (for example, in the case of alcoholic cirrhosis). In cirrhosis of viral etiology, the administration of a-interferon often causes an increase in PE, which limits its use. However, in case of cirrhosis as a result of HBV infection, the nucleotide analogue lamivudine, which suppresses the activity of the virus and, as a result, improves the functional state of the liver, can be successfully used. 2. Elimination of trigger and aggravating factors of PE: stopping bleeding, correction of hypovolemia, maintaining acid-base and electrolyte balance, eliminating infection, etc. 3. Intestinal sanitation. Colon cleansing aims to remove nitrogen-containing substances, which is especially important in cases of gastrointestinal bleeding, protein overload and constipation. The use of high enemas is effective, allowing you to cleanse the large intestine to its maximum extent, right up to the blind. This can be achieved by changing the position of the patient's body: the introduction of the solution begins with the patient on the left side, then continues in the position on the back with an elevated pelvis and ends in the position on the right side. The total volume of the administered solution should be at least 1000 ml twice a day. Sodium acetate buffer (pH 4.5) or lactulose (300 ml per 700 ml of water) are used as solutions. Oral laxatives for severe PE are best administered through a nasogastric tube. Infusion of 1000 ml of 10% mannitol solution over 60–90 minutes, causing osmotic diarrhea, ensures almost complete elimination of intestinal contents over the next 3–4 hours. In case of gastrointestinal bleeding, mannitol should be administered through a tube until a clear solution is released from the rectum. In addition to mannitol, 20–30% magnesium sulfate (50–100 ml), as well as a complex solution consisting of sodium bicarbonate, sodium chloride, potassium chloride, sodium sulfate and polyethylene glycol, can be used. 4. Diet. In patients with any stage of PE, it is advisable to limit protein intake from food. For latent PE, it is recommended to limit protein to approximately 40 g/day (0.6 g/kg body weight), at stages I–II – to 30 g/day (0.4 g/kg). At stages III–IV, it is optimal to transfer the patient to tube and parenteral nutrition with a protein content of about 20 g/day. Protein in the diet of a patient with PE should be represented mainly by proteins of plant origin and lactalbumin due to their better tolerability. Plant proteins are richer in ornithine and arginine and contain less methionine and aromatic amino acids. In some cases, meat, fish, eggs and cheeses should be completely excluded from the diet. At the same time, long-term and sharp protein restriction promotes the breakdown of endogenous proteins, which leads to an increase in the concentration of nitrogen-containing compounds in the blood, so the intake of dietary protein after improvement of the condition should be increased by an average of 10 g every 3 days. After eliminating signs of PE (including the results of psychometric tests), the daily amount of protein can be increased to 80–100 g/day (1–1.5 g/kg). It must be taken into account that each patient with cirrhosis has his own threshold for tolerance to dietary protein, and in case of protein intolerance, its deficiency must be replenished with parenteral infusions. The calorie content of food (1800–2500 kcal/day) is ensured by an adequate intake of fats (70–140 g) and carbohydrates (280–325 g). Carbohydrates help reduce the concentration of ammonia and tryptophan in plasma, however, it should be remembered that patients with cirrhosis tend to develop impaired glucose tolerance, which sometimes requires the administration of insulin. The use of fructose, sorbitol and xylitol is inappropriate due to the increased risk of lactic acidosis. The food of a patient with cirrhosis must also contain adequate amounts of vitamins and microelements; If the absorption of fat-soluble vitamins is impaired, parenteral administration is indicated. Drug therapy Lactulose. Being a non-absorbable disaccharide (1,4-b-galactoside-fructose), lactulose reduces the intestinal pH, prevents the proliferation of ammonium-producing bacteria, the absorption of ammonia and amino-containing compounds, and the breakdown of glutamine in the intestinal mucosa. Lactulose is prescribed 2-3 times a day, the dosage of the drug is individual; An increase in stool frequency to 2-3 times a day is considered a simple but reliable criterion for effectiveness. The onset of this effect reflects a decrease in colon pH < 6.0. Side effects of lactulose include nausea, vomiting, loss of appetite, bloating, diarrhea, and tenesmus. Prolonged diarrhea can lead to dehydration and electrolyte imbalance, so when selecting a dose, you should adhere to the rule regarding stool frequency: “no less than two, no more than three.” Some patients do not tolerate lactulose well due to its sweet taste; To improve the taste, it is recommended to add lemon juice. Intermediate metabolites of the urea cycle. This group includes L-ornithine-L-aspartate, L-ornithine-a-ketoglutarate and arginine malate. The most widely used L-ornithine-L-aspartate is available both in the form of a solution for intravenous infusion and in the form of a granulate for oral administration. The mechanisms of action of the drug are based on: 1) stimulation of enzymes involved in ammonia detoxification - ornithine carbamoyl transferase and glutamine synthetase; 2) the inclusion of ornithine and aspartate as substrates in the ornithine Krebs urea cycle (the formation of urea from ammonia). The standard regimen of use involves intravenous drip administration of 20–30 g of the drug for 7–14 days, followed by transition to oral administration of 9–18 g per day. With long-term use (6-month course of 9 g per day orally), the drug effectively prevents relapses of PE. Sodium benzoate and phenylacetate have been used successfully to treat patients with congenital defects in ornithine cycle enzymes. The ability of drugs to bind ammonia in the blood serves as the basis for its use in patients with PE, but their effect is short-lived. Branched chain amino acids (BCAA). Although amino acid imbalance is considered as one of the pathogenetic factors of PE, no correlation has been found between the ratio of aromatic amino acids and ACC, on the one hand, and the severity of PE, on the other. Probably, the positive clinical effect of ACC infusions is due to a decrease in protein catabolism in the liver and muscles, as well as an improvement in metabolic processes in the brain. It should be taken into account that ACC are an important source of protein for patients with PE who require dietary protein restriction. Recommended dosage is 0.3 g/kg/day. Antibiotics are prescribed to suppress ammonium-producing intestinal microflora. Neomycin (dose 6–8 g/day) and paromocycin (3–4 g/day) are currently rarely used due to their oto- and nephrotoxicity. In recent years, preference has been given to the safer rifaximin (daily dose 1200 mg, course duration 1–2 weeks). Other antibiotics used in patients with PE include ciprofloxacin, metronidazole, and rifaximin. When considering the possibility of prescribing antibiotics to patients with PE, it is important to consider not only their side effects, but also the cost/effectiveness ratio. It can be stated that the therapeutic niche of antibiotics for PE is represented by two groups of patients: those in need of enhancing the effect of lactulose; unable to tolerate standard therapy. Flumazenil, a benzodiazepine receptor antagonist, is prescribed intravenously in a bolus at a dose of 0.2–0.3 mg, then in a drip of 5 mg/hour. After the patient’s condition improves, the patient switches to oral administration at a dose of 50 mg/day. The effectiveness of the drug is higher if the cause of the manifestation of PE was the use of barbiturates or benzodiazepines.
Literature 1. Ivashkin V.T., Nadinskaya M.Yu., Bueverov A.O. Hepatic encephalopathy and methods of its metabolic correction. Bol. org. digestive 2001; 3:25–7. 2. Lebezev V.M., Gubsky L.V. Hepatic encephalopathy during surgical treatment of patients with portal hypertension. Wedge. honey. 1995; 73:37–9. 3. Conn HO, Bircher J. (eds.). Hepatic encephalopathy: syndromes and therapies. Bloomington, Illinois: Medi–Ed Press 1994: 243 p. 4. Hepatic encephalopathy. In: Kuntz E., Kuntz H.–D. Hepatology. Principles and practice. Springer 2002: 234–54. 5. Plauth M., Egberts E.–H. Was ist gesichert in der therapy der hepatischen enzephalopathie? International 1993; 34: 35–42. 6. Stauch S., Kircheis G., Adler G. et al. Oral L–ornithine–L–aspartate therapy of chronic hepatic encephalopathy: results of placebo–controlled double–blind study. J. Hepatol. 1998; 28:856–64.